


Eighty per cent of a typical old age psychiatrist's time is spent in the assessment and management of dementia. Most of this is due to Alzheimer's disease or a combination of Alzheimer's disease and vascular dementia. Because of increased public awareness of advances in genetics and the efforts of advocacy groups in explaining the latest research findings, relatives of people with Alzheimer's disease are increasingly asking the question, “ What is my risk and what is my children's risk of developing Alzheimer's disease?”. The aim of this article is to summarise the relevant research and to suggest ways in which this can be answered.
AUTOSOMAL DOMINANT FAMILIAL ALZHEIMER'S DISEASE
Most of the highly publicised advances in the genetics of Alzheimer's disease have concerned those very rare families with autosomal dominant forms of the disease, so-called familial Alzheimer's disease (FAD) kindred, in which 50% of each generation, regardless of gender, succumb to Alzheimer's disease, usually by early mid-life (reviewed by Reference SelkoeSelkoe, 1999). The identification of causative mutations within the amyloid precursor protein (APP) and presenilin genes (PSEN1 and PSEN2) has probably overinflated the public's perception of the role of genes in causing the far more common forms of Alzheimer's disease that do not show autosomal dominant transmission. Yet, it is important to be able to recognise FAD because it is possible to offer such kindreds definitive genetic counselling, provided that the causative mutation can be identified. Furthermore, a study by Campion et al (Reference Campion, Dumanchin and Hannequin1999) suggests that FAD may occur at a frequency of around 40 per 100 000 persons at risk, which implies that there are some 600 affected individuals to be found in England and Wales, a number that suggests that a psychiatrist or neurologist will encounter an individual from a FAD pedigree at some time in their working life.
SUGGESTED APPROACH FOR HIGHLY FAMILIAL FORMS OF ALZHEIMER'S DISEASE
The function of the psychiatrist is straight-forward. The first step is to confirm that the diagnosis is actually one of Alzheimer's disease and not one of the other, often familial, dementias such as Huntington's disease, frontal-temporal dementias, Creutzfeldt—Jakob disease (CJD) or cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL). Next, the clinician should take as detailed a family history as possible in order to try to determine whether the sufferer comes from a pedigree with autosomal dominant FAD: the occurrence of numerous individuals with early-onset disease (<55 years) in several generations is highly suggestive. The presence of many affected individuals in one generation may also raise suspicion because it should not be forgotten that early death from other causes might prevent manifestation of the disorder in carriers of a disease gene. In practice, we recommend that families containing three members with a history of early-onset Alzheimer's disease occurring before the age of 60 years should, if they request advice, be referred to a clinical geneticist. Some forms of FAD linked to PSEN2 mutations occur after the age of 65 years, and so the psychiatrist should have a low threshold for discussing a pedigree with a clinical geneticist.
Predictive or prenatal testing is potentially feasible in families with FAD but will depend upon the availability of DNA from at least one affected individual in order to establish which mutation is causing disease in that family. It is worth stating that genetic counselling is a highly specialised area and it is strongly recommended that psychiatrists do not involve themselves without the appropriate training.
The study by Campion et al (Reference Campion, Dumanchin and Hannequin1999) analysed DNA from 34 families with FAD (obtained from all over France) for causative mutations. Probably pathogenic mutations were found in the PSEN1 gene in 19 families, mutations within the APP gene in another 5 and no mutations in the PSEN1, PSEN2 or APP genes in the remaining 10 families. Thus, individuals from FAD pedigrees need to be informed that there is a 30% chance that, with the current state of our knowledge, no causative mutation will be found. If no mutation is found then, if the pedigree is strongly suggestive of an autosomal dominant disorder, the geneticist may choose to go on to search for mutations in genes associated with similar conditions — prion protein (PrP) in CJD and tau in frontal—temporal dementias, for example. Yet another factor to consider is that there has been one report of apparent non-penetrance of a PSEN1 mutation in a healthy 68-year-old member of a FAD pedigree (Reference Rossor, Fox and BeckRossor et al, 1996). Notwithstanding these caveats, in the absence of any preventive treatment, the take-up for predictive testing is likely to be low, as has been the case in Huntington's disease (Reference Binedell, Soldan and HarperBinedell et al, 1998).
SUGGESTED APPROACH FOR NON-MENDELIAN ALZHEIMER'S DISEASE
The great majority of cases of Alzheimer's disease do not result from disease-causing mutations in a single gene. Here inheritance does not follow simple Mendelian ratios but appears to reflect the presence of a number of different genetic risk factors together with environmental factors.
When faced with someone who is worried about their risk of developing Alzheimer's disease it is often worth pointing out that we are all at some risk of developing Alzheimer's disease provided that we live long enough. The large Rotterdam study suggests that the risk at age 55 years of developing dementia in the following 35 years is 0.26 for a woman and 0.15 for a man (Reference Ott, Bretelar and van HarskampOtt et al, 1998). In this study 73% of dementia was wholly or partially attributable to Alzheimer's disease. The large continuing Framingham study yields somewhat lower lifetime risks of Alzheimer's disease after the age of 65 years: 6.3% for men and 12% for women, with a corresponding risk for all dementia of 10.9% and 19%, respectively (Reference Seshadri, Wolf and BeiserSeshadri et al, 1997). Many epidemiological studies suggest that women are at increased risk of Alzheimer's disease. The reasons for this are not clear, but increased longevity compared to men, increased survival with the disease and some increase in intrinsic vulnerability probably all play a part.
Prevalence studies tell us that dementia doubles every 5 years after the age of 65 years up to the age of around 85 years, after which time the rate of increase appears to slow (Reference Heeren, Lagaay and HijmansHeeren et al, 1991; Reference Skoog, Nilsson and PalmertzSkoog et al, 1993; Reference Ritchie and KildeaRitchie & Kildea, 1995; Reference Breitner, Wyse and AnthonyBreitner et al, 1999; Reference von Strauss, Viltanen and De Ronchivon Strauss et al, 1999). It is likely that some of this slowing is due to very old people not living as long with their dementia as younger sufferers do, although it is also possible that survivors into late old age are relatively resistant to developing dementia (Reference DrachmanDrachman, 1994). Prevalence data are summarised in Table 1.
Table 1 Prevalence of dementia by age (from Reference Ritchie and KildeaRitchie & Kildea, 1995, with permission)
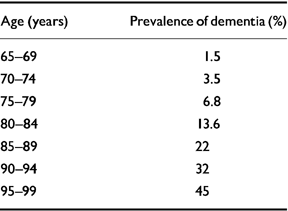
| Age (years) | Prevalence of dementia (%) |
|---|---|
| 65-69 | 1.5 |
| 70-74 | 3.5 |
| 75-79 | 6.8 |
| 80-84 | 13.6 |
| 85-89 | 22 |
| 90-94 | 32 |
| 95-99 | 45 |
In fact, two nearly completely ascertained community-based studies, one from Japan and one from The Netherlands, found that the prevalence of dementia in centenarians was very high: 70% in the Japanese and 88% in the Dutch sample (Reference Asada, Yamagata and KilnoshitaAsada et al, 1996; Reference Blansjaar, Thomassen and van SchaickBlansjaar et al, 2000). Asada et al (Reference Asada, Yamagata and Kilnoshita1996) attributed some 76% of the dementia to Alzheimer's disease and demonstrated that the 6-month mortality rate was 27% for the centenarians with dementia, whereas none of the non-demented centenarians died.
Thus, prevalence figures for dementia tell us that many of us are at some risk of developing dementia, provided that we live long enough. This is obviously too simplistic: most people do not expect to live into their mid-90s; a much more reasonable question is, “What is my chance of developing Alzheimer's disease by the age of 85 years?”. Family studies in Alzheimer's disease can go some way towards allowing us to answer this question for the relatives of affected individuals.
FAMILY STUDIES AND RISK OF DEMENTIA
In the 1980s and early 1990s there were a number of family studies that attempted to identify the inheritance pattern of Alzheimer's disease and to quantify the risk to first-degree relatives in families with a history of Alzheimer's disease, in comparison with family members of control individuals (summarised in Reference McGuffin, Owen and O'DonovanMcGuffin et al, 1994). There have also been a few studies based on more representative community samples.
Overall, the findings from studies based in memory clinics (that are likely to be centres of secondary referral) suggest that 30-48% of probands with Alzheimer's disease have a history of affected first-degree relatives compared with 13-19% of controls. This translates to 6-14% of the relatives of patients with Alzheimer's disease having a history of Alzheimer's disease compared with 3.5-7% of the first-degree relatives of healthy controls. Kaplan—Meier life table analysis has been used in these studies to infer that the cumulative risk of dementia by age 90 varies between 30 and 50% compared with between 10 and 23% in control relatives. However, as has been pointed out by Breitner, owing to competing causes of death, only about one-third of this theoretical familial predisposition to Alzheimer's disease is realised in the usual life span. This translates to an actual predicted risk of developing Alzheimer's disease in the first-degree relatives of probands with Alzheimer's disease of 15-19%, compared with 5% in controls (Reference Breitner, Murphy and SilvermanBreitner et al, 1988; Reference BreitnerBreitner, 1991).
The latest systematically ascertained twin studies on the heritability of Alzheimer's disease also tend to support the estimates of familial risk derived from family studies. Probandwise, concordance rates of about 40% and 84%, respectively, are seen in fraternal and identical twins (Reference Bergem, Engedal and KringlenBergem et al, 1997). Because fraternal twins are genetically equivalent to ordinary first-degree relatives, the observed morbid risk of developing Alzheimer's disease is similar to that estimated for first-degree relatives in family studies.
A study by Silverman et al (Reference Silverman, Ge and Zaccarto1994) suggested that, in large measure, the familial component of risk to the relatives of probands with Alzheimer's disease was expended by the end of the ninth decade, after which time the risk was very similar to that in controls. A number of other family history studies have suggested that familial factors are more prominent when onset is earlier (Reference McGuffin, Owen and O'DonovanMcGuffin et al, 1994). Thus, much of Alzheimer's disease in late old age may be considered as not very familial and merely an expression of one of the ways in which the ageing process is manifested in those few survivors into late old age.
The findings from these family studies can be used to advise relatives in only the broadest terms. One can do little more than to say that, on balance, the risk to the first-degree relatives of patients with Alzheimer's disease who developed the disorder at any time up to the age of 85 years is increased some threefold to fourfold relative to the risk in controls. This would seem to translate to a risk of developing Alzheimer's disease of between one in five and one in six, which, although an improvement on a risk of one in two, can still be rather frightening. In order to put this risk into better perspective, and to help allay the client's anxiety, it may be preferable to present the risk estimates in a graphical form, as is done in Fig. 1 (from Reference Breitner, Murphy and SilvermanBreitner et al, 1988). If this is done, clients can see that even at 78 years, the age of greatest risk, the actual predicted risk is only some 3% and that the cumulative risk (half the area under the curve f(x)·S(x)) from age 65 to 78 is probably less than 10%. In our experience many tell us that this is reassuring — partly because their worries were about getting Alzheimer's disease in their fifties “because this is when familial Alzheimer's disease happens” or in their sixties/seventies, thus preventing them from enjoying retirement.
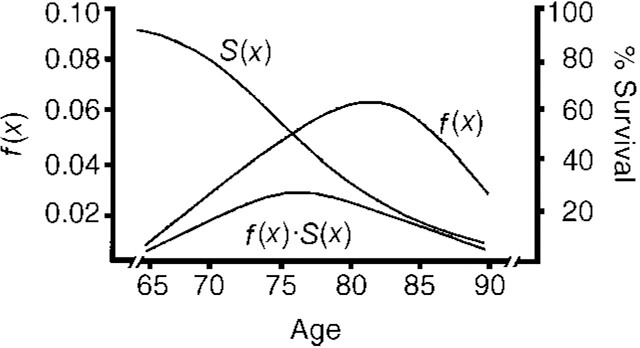
Fig. 1 f(x) represents the probability of a hypothetical relative of a patient with Alzheimer's disease developing Alzheimer's disease at age x in the absence of any competing form of mortality. S(x) represents the actuarial probability of surviving to age x. The product of f(x)·S(x) represents the probability of the relative surviving to age x and developing Alzheimer's disease. Adapted from Breitner et al (Reference Breitner, Murphy and Silverman1988)
In the case of patients with Alzheimer's disease who became demented late in old age, say by their 80s, relatives probably run the same 30-50% risk of developing dementia as anyone else who lives to the age of 90 years and beyond. Of course, it is possible that some of the same genes that confer longevity may also increase the risk of developing Alzheimer's disease. Like other disorders that reflect the combined action of several genes, the risk to relatives drops rapidly as the degree of genetic relatedness falls. Data are limited but the risk to second-degree relatives, such as grand-children, is probably less than twice the population levels (Reference Heston, Mastri and AndersonHeston et al, 1981).
THE APOLIPOPROTEIN E EFFECT
Apolipoprotein E (apoE) is a protein with roles in lipid metabolism and tissue repair. Its primary site of biosynthesis is the liver, but the second major site of synthesis is the brain. Like APP, the synthesis of apoE is up-regulated after the nervous system has been damaged. There are three commonly occurring polymorphic forms of apoE, known as apoE2, apoE3 and apoE4, which originate from the APOE ϵ2, APOE ϵ3 and APOE ϵ4 alleles of the gene. The key observation, originally made by Strittmatter et al (Reference Strittmatter, Saunders and Schmechel1993), is that the frequency of APOE ϵ4 in patients with Alzheimer's disease (namely 0.3-0.5) is greater than the APOE ϵ4 frequency of 0.1-0.15 in age-matched or population controls. Some studies suggest that the APOE ϵ2 allele is under-represented in Alzheimer's disease and may, by inference, be protective (reviewed by Reference Farrer, Cupples and HainesFarrer et al, 1997). Consistent with this idea is the finding that healthy centenarians have a higher APOE ϵ2 frequency than general population controls (Reference Schächter, Faure-Delanef and GuénotSchächter et al, 1994; Reference Kehoe, Russ and McIlroyKehoe et al, 1999).
The APOE ϵ4 association with Alzheimer's disease has been replicated in many laboratories around the world. A meta-analysis summarised the data and demonstrated that the association was found in people of European, African American, American Hispanic and Japanese origin (Reference Farrer, Cupples and HainesFarrer et al, 1997). Exactly how APOE ϵ4 and the apoE4 protein influence the pathophysiology of Alzheimer's disease is still unknown, but bearers of this risk allele appear to get Alzheimer's disease earlier and develop a heavier amyloid burden.
CLINICAL SIGNIFICANCE OF APOLIPOPROTEIN E GENOTYPE
Most studies show that patients with Alzheimer's disease having APOE ϵ 4 deteriorate no more rapidly than those without this allele (Reference Corder, Saunders and StrittmatterCorder et al, 1995; Reference Growdon, Locascio and CorkinGrowdon et al, 1996; Reference Holmes, Levy and McLoughlinHolmes et al, 1996; Reference Kurz, Egensperger and HauptKurz et al, 1996; Reference Stern, Brandt and AlbertStern et al, 1997). It is possible that APOE ϵ4 is a marker for a poor response to treatment with acetylcholinesterese inhibitors, particularly, perhaps, in women (Poirer et al, 1995; Reference Farlow, Lahiri and PoirerFarlow et al, 1998).
The consensus of opinion appears to be that APOE genotype determines ‘when’ rather than ‘whether’ one succumbs to Alzheimer's disease. For example, the Cache County study (Reference Meyer, Tschanz and NortonMeyer et al, 1998, Reference Breitner, Wyse and AnthonyBreitner et al, 1999) on an elderly population of almost 5000 found 22 cases of Alzheimer's disease among 141 APOE ϵ4 homozygotes, 118 among 1452 APOE ϵ4 heterozygotes and 80 among 3339 not bearing the APOE ϵ4 allele. There appeared to be a plateau in each group's survival curve beyond which no new cases of Alzheimer's disease were seen. For APOE ϵ4 homozygotes no new cases were seen after the age of 84 years, with nine individuals surviving without Alzheimer's disease for a combined total of a further 37 years. For APOE ϵ 4 heterozygotes the last onset of Alzheimer's disease was at 99 years, with four long-term survivors. The last onset of dementia in individuals without the APOE ϵ4 allele was at age 95 years, with 31 surviving free of dementia for a combined total of 100 years thereafter. This differential effect of APOE genotype on the age of maximum risk of Alzheimer's disease is also suggested in the APOE meta-analysis of Farrer et al (Reference Farrer, Cupples and Haines1997) and also in the study by Asada et al (Reference Asada, Yamagata and Kilnoshita1996).
A Bayesian analysis has been used by Seshadri et al (Reference Seshadri, Drachman and Lippa1995) to relate the APOE genotype to the lifetime risk of developing Alzheimer's disease. When the estimates of dementia risk derived from the Framingham study (Reference Seshadri, Wolf and BeiserSeshadri et al, 1997) are substituted in the analysis, somewhat lower risk estimates, which are stratified according to gender, are obtained (Table 2).
Table 2 Remaining lifetime risk at age 65 years of developing Alzheimer's disease according to gender and apolipoprotein E (APOE) status
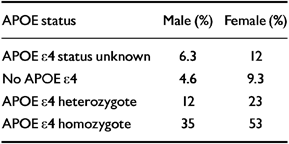
| APOE status | Male (%) | Female (%) |
|---|---|---|
| APOE ϵ4 status unknown | 6.3 | 12 |
| No APOE ϵ4 | 4.6 | 9.3 |
| APOE ϵ4 heterozygote | 12 | 23 |
| APOE ϵ4 homozygote | 35 | 53 |
These estimates suggest that, at most, some 50% of APOE ϵ4 homozygotes will develop Alzheimer's disease within their lifetime. These predictions agree well with the general population-based study of Henderson et al (Reference Henderson, Easteal and Jorm1995), which suggested that the risk of developing dementia by age 90 years in APOE ϵ4 homozygotes was about 50%.
POSSIBLE CLINICAL USES OF APOE GENOTYPING IN ALZHEIMER'S DISEASE
From the foregoing it is obvious that knowledge of a person's APOE genotype is of little more use in predicting their chances of succumbing to Alzheimer's disease than is knowledge of their family history of dementia. It seems that even individuals with the APOE ϵ4/APOE ϵ4 genotype have, on average, a greater than 50% chance of escaping the disease. Therefore, APOE genotyping currently has no role in predicting the risk of developing Alzheimer's disease.
Some claims have been made that APOE genotyping may be an aid to diagnosing Alzheimer's disease. The study by Mayeux et al (Reference Mayeux, Saunders and Shea1998) suggested that for patients referred to specialised assessment centres for Alzheimer's disease, APOE genotyping used in combination with clinical criteria might improve the specificity of the diagnosis. The data show that whereas the demonstration of one or more APOE ϵ4 allele in a person suspected of suffering with dementia slightly increases the accuracy of a clinical diagnosis of Alzheimer's disease, the absence of an APOE ϵ4 allele has little value in either endorsing or refuting a clinical diagnosis of Alzheimer's disease. Thus, even in a selected group of patients, presumably all with a high a priori chance of Alzheimer's disease, APOE genotyping seems to confer negligible diagnostic benefit.
GENETIC COUNSELLING AND ALZHEIMER'S DISEASE
A number of groups in the UK and in the USA have formulated consensus guidelines regarding genetic testing and genetic counselling in Alzheimer's disease, including Alzheimer's Disease International (Reference Brodaty, Conneally and GauthierBrodaty et al, 1996), the American College of Medical Genetics/American Society of Human Genetics Working Group on APOE and Alzheimer's Disease (Reference Farrer, Brin and ElsasFarrer et al, 1995) and the Alzheimer's Association/National Institute of Aging (Reference Davies, Gilman and GrowdonDavies et al, 1998). In the UK, the Alzheimer's Disease Genetics Consortium meets regularly to discuss the issues surrounding the clinical use of genetics in Alzheimer's disease. These issues include not only whether to test, but how to counsel patients and issues regarding confidentiality and consent, research ethics and concerns regarding insurance (Reference Tunstall and LovestoneTunstall & Lovestone, 1999). The various groups that have drawn up the following consensus guidelines are generally of one mind with regard to the clinical response to our growing understanding of the genetics of Alzheimer's disease.
-
(a) Families where there is evidence of FAD (or indeed any familial early-onset dementia) should be referred to a specialist centre — in the UK this will be a regional genetics department.
-
(b) Counselling for such families should follow the process established for Huntington's disease.
-
(c) For late-onset Alzheimer's disease all the groups are agreed that there is no role for APOE genotyping in prediction or risk assessment.
RECOMMENDATIONS
Apparent FAD
Early-onset familial dementia is straight-forward — refer to a regional genetics department.
Late-onset Alzheimer's disease
For late-onset Alzheimer's disease there is wide agreement that there is no genetic test available, but genetic testing is not the same as genetic counselling. The absence of a test should not mean that relatives are denied information and the opportunity to discuss their concerns. So, when asked “ am I likely to get Alzheimer's disease?” by the relative of a patient with late-onset Alzheimer's disease, how should one answer? In most cases of Alzheimer's disease it is only possible to advise relatives of their risk in the broadest terms. Extrapolating from family history studies, it is possible to say that the risk to the children is in the region of one in five to one in six and one should be prepared to illustrate what this means in an easy to understand graphical form. We believe that it is permissible for such a broad estimate of risk to be imparted by a consultant or doctor not specifically trained in genetic counselling.
Occasionally, psychiatrists encounter families with several siblings affected by late-onset Alzheimer's disease and a history of dementia in previous generations. Such families almost certainly exhibit high genetic loading, but there are few reliable data on whether and to what extent risk increases with the number of affected relatives, although studies of other common disorders suggest that this is likely to be the case. However, it should be borne in mind that the number of affected relatives may well depend on factors affecting longevity as well as on the degree of genetic loading for Alzheimer's disease, and it may not be advisable or possible to base discussion on risk estimates that are increased when several relatives have been affected in late old age. There is also preliminary evidence for substantially increased risk in the offspring of parents who both have a diagnosis of Alzheimer's disease (Reference Bird, Nemens and KukullBird et al, 1993). In such pedigrees, if relatives request further information, it may be worth taking a detailed family history and consulting with a clinical geneticist.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
▪ The taking of a family history to look for other cases of dementia in first-degree relatives should be part of every routine dementia diagnostic process.
-
▪ A clinician should be able to recognise possible familial Alzheimer's disease and be able to refer the family on to a regional genetics centre if members of the family request genetic counselling.
-
▪ A clinician should be able to advise relatives from less highly genetically loaded pedigrees with Alzheimer's disease about their risk in broad terms and seek advice from a regional genetics centre if further clarification is required.
LIMITATIONS
-
▪ In the interests of space and clarity, citation of the literature is selective.
-
▪ Estimates of risk are in some instances approximate and may need to be modified in the light of future work.
-
▪ Few data relating to vascular and mixed forms of dementia are available.




eLetters
No eLetters have been published for this article.