


Clinical characteristics
The American Psychiatric Association in 1980 first defined generalised anxiety disorder (GAD) as a condition characterised by worry combined with somatised anxiety. Subsequent revisions of the DSM nosology have focused on a chronic state of worry combined with muscular tension, cognitive dysfunction and poor sleep. Large-scale epidemiological studies indicate a lifetime prevalence of 5-6% (Reference Wittchen, Zhao and KesslerWittchen et al, 1994). It is often comorbid with other disorders, particularly major depression, panic disorder, phobic states and substance misuse (Reference Merikangas, Angst and EatonMerikangas et al, 1996; Reference Judd, Kessler and PaulusJudd et al, 1998). It is associated with psychosocial impairment, increased morbidity and mortality and therefore constitutes a public health problem (Reference Kessler and WittchenKessler & Wittchen, 2001).
Current treatments
Although the benzodiazepines are used as anxiolytics in many conditions, they have not been indicated specifically for GAD. In the 1980s, buspirone was introduced for treating GAD-related symptoms (Reference Goa and WardGoa & Ward, 1986). Supportive therapy, muscle relaxation and cognitive—behavioural techniques have some evidence of effectiveness for up to 6 months (Reference Fisher and DurhamFisher & Durham, 1999).
Study rationale
The efficacy in panic anxiety of clomipramine, enhancing both serotonergic and noradrenergic transmission, and the efficacy of imipramine and trazodone in GAD called for a study of venlafaxine in patients with anxiety disorder (Reference Modigh, Westberg and ErikssonModigh et al, 1992; Reference Rickels, Downing and SchweizerRickels et al, 1993). This study was designed to compare the short— and long-term efficacy and safety of three fixed doses of the serotonergic and noradrenergic reuptake inhibitor (SNRI) venlafaxine extended release (ER) with placebo in out-patients with GAD.
METHOD
Patient population
A multi-centre, double-blind, randomised, parallel-group design was used at a total of 55 sites in Belgium, Finland, France, Sweden and the UK (see Appendix). Primary care and psychiatric out-patients aged at least 18 years were recruited if they met DSM-IV (American Psychiatric Association, 1994) criteria for GAD and had given signed informed consent. The diagnosis requires that patients have had symptoms of excessive anxiety and worry that were difficult to control for most days during the past 6 months. In addition, the anxiety and worry were associated with at least three of six symptoms — being restless/keyed-up, being easily fatigued, having difficulty concentrating or having one's mind go blank, and experiencing irritability, muscle tension or sleep disturbance. All patients were to have minimum scores of 20 on the Hamilton Rating Scale for Anxiety (HRSA; Reference HamiltonHamilton, 1959) and scores ≥ 2 on item 1 (anxious mood) and item 2 (tension). All the participating physicians were trained in making the diagnosis and in rating the symptoms using the HRSA in joint sessions, including a test and validation of interrater reliability.
Patients were excluded from the study if they had had a major depressive disorder in the previous 6 months or exhibited clinically significant depressive symptomatology. In addition, they were excluded if they had any clinically important medical disease or abnormality on physical examination as well as other psychiatric disorders, excessive consumption of caffeine-containing food and drink and use of pharmacological or non-pharmacological drugs with psychotropic effects as checked with a drug screen. Maintenance medications that were not psychopharmacological but had psychotropic effects, such as beta-blockers for hypertension, were permitted.
Study design
After a 4-10-day single-blind placebo washout period, the study consisted of a 24-week double-blind treatment period followed by a 1-week single-blind placebo discontinuation period. The patients were assigned randomly to receive one of the three non-titrated fixed doses of venlafaxine ER (37.5, 75 or 150 mg) or placebo once daily in the morning for up to 24 weeks. A randomisation schedule in blocks of four was generated for packaging and labelling by the Biostatistics Section of Wyeth-Ayerst Research. The 75-mg and 150-mg dosages of venlafaxine ER were chosen on the basis of the usual dosage recommendations for the treatment of depression. The lower dose of 37.5 mg of venlafaxine ER was included to evaluate the dose-response relationship and the minimal effective dose in anxiety.
Assessments
The following rating scales were completed at baseline and at each visit (weeks 1, 2, 3, 4, 6, 8, 10, 12, 16, 20, 24 and 25): the HRSA; the Hospital Anxiety and Depression (HAD; Reference Zigmond and SnaithZigmond & Snaith, 1983) scale (a 14-item self-administered measure); the Brief Scale for Anxiety (BSA; Reference Tyrer, Owen and CicchettiTyrer et al, 1984); and the Clinical Global Impression of Improvement (CGI-I; Reference GuyGuy, 1976). Changes in social functioning were assessed using the Self-Rated Social Adjustment Scale (SAS-SR; Reference Weissmann and BothwellWeissman & Bothwell, 1976) at baseline and at weeks 8 and 24. Tolerability was assessed by spontaneous replies to an open question at each visit. Safety was assessed by means of a physical examination at screening, by monitoring weight, blood chemistry and blood pressure and by assessments of the resting electrocardiogram (ECG). The Physician Withdrawal Checklist (PWC; Reference Rickels, Schweizer and CaseRickels et al, 1990) was administered at screening, baseline and weeks 24 and 25. An assessment of compliance was made by counts of returned medication at each visit.
Data management and statistics
The statistical analyses were based on the pooled data from all study sites. Data from centres with small sample sizes were combined with other centres before the study blind was broken, reducing it to 14 centre groups: seven in the UK, three in France, two in Sweden and one each in Belgium and Finland. The main efficacy analyses considered the intention-to-treat population, which included all patients who had received at least one dose of study medication and who had a baseline and at least one on-therapy efficacy assessment.
Outcome variables
The end of week 8 was considered the primary time point for short-term treatment and the end of week 24 for long-term treatment, but data for assessments at other weeks are also described. The primary outcome variables for the assessment of efficacy were defined a priori as the HRSA total, the HRSA psychic anxiety factor, the HAD anxiety sub-scale and the CGI-I rating. The comparisons of principal interest were between each dose of venlafaxine ER and placebo for these variables. All other comparisons and all other variables were considered secondary. Data were analysed using both the last observation carried forward (LOCF) method and the observed data at each time point. ‘Response to treatment’ was defined as a decrease of at least 50% in the HRSA total score from baseline or a CGI-I score of 1 or 2 (much or very much improved).
Statistical analyses
For the primary variables of interest, a Bonferroni correction for multiple testing was made. Differences were considered significant for primary pairwise comparisons when P values were ≤0.05 for the global F-test and ≤0.017 for the pairwise comparison. All other comparisons were considered significant if the P value of a pairwise comparison was ≤0.05 for both the global F-test and the pairwise comparison. All hypothesis testing was two-sided. The HRSA total and factor scores and the HAD sub-scales were analysed with a two-way analysis of covariance (ANCOVA), with treatment, centre and their interaction as factors in the model and with the baseline value as covariate. The CGI-I was analysed by using a two-way analysis of variance (ANOVA), with treatment, centre and their interaction as factors. Because of the low numbers of patients in some treatment groups at the later time points for the observed case analyses and because no evidence of a treatment by centre interaction was found in the LOCF analysis, the interaction term was dropped from the model to allow the adjusted means to be estimated for the observed case analysis only. The results described used ‘adjusted means’ derived from ANOVA/ANCOVA. ‘Visit-minus-baseline’ change scores were used in the appropriate statistical model in all cases except the CGI-I, where the actual scores were used. Responder rates at each time point were compared by means of Fisher's exact test. Withdrawal was assessed by means of ANCOVA on the PWC total score at the withdrawal assessment.
Safety
All patients assigned to double-blind treatment were included in the evaluation of safety and tolerability. Adverse events were coded using COSTART (Food and Drug Administration, 1989), by body system and preferred term. Laboratory data and vital signs were assessed using group mean changes and criteria for ‘potentially clinically important change’ that identified values or changes that fell outside predetermined limits.
Protocol violations
By convention, the ITT population includes patients who are found to violate the inclusion criteria. In this study, 25 subjects reported a current illness duration of less than 6 months, although they met all the other diagnostic criteria for GAD. The results remained unchanged when these subjects were excluded from the analysis.
RESULTS
Baseline characteristics
A total of 541 patients were assigned to treatment and 529 qualified for inclusion in the ITT analysis. The other 12 patients had no primary efficacy evaluations during therapy. Prospective randomisation at baseline resulted in groups that were similar with respect to age, male/female ratio, height, weight, duration of current illness, baseline HRSA total and psychic anxiety factor scores, and the HAD anxiety subscale. Patients were predominantly female, with a mean age in the mid-forties, a mean GAD episode duration of 10 years and a mean severity score on the HRSA of 26-27 (Table 1).
Table 1 Summary of demographic characteristics

| Population subset | Placebo | Venlafaxine ER | ||
|---|---|---|---|---|
| 37.5 mg | 75 mg | 150 mg | ||
| Intent-to-treat (n=529) | 130 | 138 | 130 | 131 |
| Safety (n=541) | 130 | 140 | 134 | 137 |
| Female n (%) | 76 (58) | 80 (57) | 83 (62) | 89 (65) |
| Male n (%) | 54 (42) | 60 (43) | 51 (38) | 48 (35) |
| Mean age (years) | 46.1 | 45.4 | 44.1 | 45 |
| Range | 18-86 | 19-79 | 18-75 | 20-82 |
| Mean duration of current illness (weeks) | 491.6 | 567 | 470.9 | 429.9 |
| Mean baseline HRSA | 26.7 | 26.6 | 26.3 | 26.3 |
| Range | 20-52 | 20-44 | 20-43 | 17-38 |
Nearly three-quarters of patients in all groups (74%; 399/541) had received prior treatment for anxiety, mostly benzodiazepines and antidepressants. Other prior treatments for anxiety that had been used by at least 10% of patients in each group included hypnotics (mostly benzodiazepines) and β-adrenergic receptor blocking agents. Eighty-five per cent (461/541) of patients received some type of non-anxiolytic concomitant therapy during the study (26 patients had beta-blocker maintenance therapy and 52 patients used zolpidem or chloral hydrate occasionally for sleep). There were no marked differences among the treatment groups in terms of the classes of concomitant medications given.
Discontinuations
A total of 147 (36%) patients discontinued treatment over the 24-week treatment period (Table 2). The number of patients who discontinued did not differ significantly among the treatment groups, but there were relatively fewer discontinuations among patients receiving the higher doses of venlafaxine ER (75 or 150 mg/day). Patients who received placebo were significantly more likely to discontinue because of lack of efficacy than patients receiving venlafaxine ER (P <0.001). The most common reason for discontinuation among patients receiving placebo or the lowest dose of venlafaxine ER (37.5 mg) was ‘unsatisfactory response/efficacy’. Among patients receiving the higher dose of venlafaxine ER, adverse events were the most common reason for discontinuation. Discontinuations due to adverse events were as common among patients receiving placebo as among those receiving any of the doses of venlafaxine ER, although there were more discontinuations on venlafaxine ER during the early weeks of treatment.
Table 2 Overview of reasons for discontinuation in intention-to-treat sample (% patients)

| Primary reason | Placebo (n=130) | Venlafaxine ER | ||
|---|---|---|---|---|
| 37.5 mg (n=140) | 75 mg (n=134) | 150 mg (n=137) | ||
| Any reason | 45 | 38 | 33 | 31 |
| Unsatisfactory response/efficacy1 | 21 | 17 | 10 | 4 |
| Adverse event | 14 | 11 | 17 | 16 |
| Other2 | 11 | 9 | 5 | 9 |
Efficacy evaluation
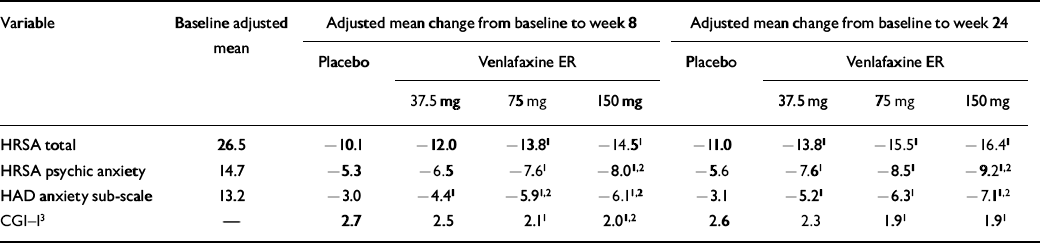
For evaluation of efficacy, the primary comparisons of interest for both the short-and long-term evaluations (up to weeks 8 and 24, respectively) are summarised in Table 3 for the LOCF analysis. In the short term, both of the higher venlafaxine ER dose groups (75 and 150 mg) showed significant differences from the placebo group on all of the primary efficacy variables. A significant difference between 37.5 mg of venlafaxine and placebo was seen only for the HAD anxiety sub-scale at week 8. The highest dose of 150 mg of venlafaxine ER also showed significantly greater efficacy than 37.5 mg of venlafaxine ER on three of the four primary efficacy variables (HRSA psychic anxiety factor, HAD anxiety, CGI-I scores) in the short term. Results after 24 weeks of treatment showed that the greater efficacy seen for the higher venlafaxine ER doses was maintained during the long term, as was the superiority over low-dose venlafaxine ER (37.5 mg). A graphic presentation of both the short— and the long-term results for the HRSA total and psychic anxiety factor is shown in Fig. 1. It is of note that similar results for 75 and 150 mg of venlafaxine ER were seen in the observed case analysis, which considers only those patients who have remained in treatment. Thus, in the observed case analysis of patients who completed the 6-month treatment, the differences from placebo in the adjusted mean change from baseline of the 75 and 150 mg doses of venlafaxine ER in the HRSA total score were 3.9 and 4.7, respectively.
Table 3 Summary of short— (week 8) and long-term (week 24) primary efficacy variables in intention-to-treat sample using the last observation carried forward method
| Variable | Baseline adjusted mean | Adjusted mean change from baseline to week 8 | Adjusted mean change from baseline to week 24 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo | Venlafaxine ER | Placebo | Venlafaxine ER | ||||||
| 37.5 mg | 75 mg | 150 mg | 37.5 mg | 75 mg | 150 mg | ||||
| HRSA total | 26.5 | −10.1 | −12.0 | −13.81 | −14.51 | −11.0 | −13.81 | −15.51 | −16.41 |
| HRSA psychic anxiety | 14.7 | −5.3 | −6.5 | −7.61 | −8.01,2 | −5.6 | −7.61 | −8.51 | −9.21,2 |
| HAD anxiety sub-scale | 13.2 | −3.0 | −4.41 | −5.91,2 | −6.11,2 | −3.1 | −5.21 | −6.31 | −7.11,2 |
| CGI—I3 | — | 2.7 | 2.5 | 2.11 | 2.01,2 | 2.6 | 2.3 | 1.91 | 1.91 |

Fig. 1 (a) Changes in the Hamilton Rating Scale for Anxiety (HRSA) total score (left) and psychic anxiety factor (right) by week in the short term (8 weeks); * P <0.05, ** P <0.01 and *** P <0.001 for venlafaxine ER v. placebo; † P <0.05 and †† P <0.01 for 150 mg venlafaxine ER v. 37.5 mg venlafaxine ER. (b) Changes in the HRSA total score (left) and psychic anxiety factor (right) by week in the long term (24 weeks); * P <0.05, ** P <0.01 and *** P <0.001 for venlafaxine ER v. placebo; † P <0.05 and †† P <0.01 for 150 mg venlafaxine ER v. 37.5 mg venlafaxine ER.
The secondary outcome variables, including social impairment, also showed a more consistent picture of efficacy in comparison with placebo for 75 and 150 mg of venlafaxine ER than for 37.5 mg of venlafaxine ER during both short— and long-term treatment (Table 4). In addition, the 150 mg dose of venlafaxine showed consistent superiority over 37.5 mg of venlafaxine ER on all secondary outcome variables except the HRSA somatic factor.
Table 4 Overview of efficacy on secondary outcome variables: adjusted means (95% CI) for intention-to-treat sample using the last observation carried forward method

| Parameter | Placebo | Venlafaxine ER | ||
|---|---|---|---|---|
| 37.5 mg | 75 mg | 150 mg | ||
| HRSA somatic anxiety factor, baseline adjusted mean 11.8 | ||||
| Week 8 | 7.0 (6.3-7.8) | 6.2 (5.5-6.9) | 5.6 (4.9-6.4)1 | 5.4 (4.6-6.1)1 |
| Week 24 | 6.4 (5.6-7.2) | 5.5 (4.8-6.3) | 4.7 (3.9-5.5)1 | 4.5 (3.7-5.3)1 |
| HRSA anxious mood item, baseline adjusted mean 2.8 | ||||
| Week 8 | 1.8 (1.6-2.0) | 1.5 (1.3-1.7)1 | 1.4 (1.2-1.6)1 | 1.2 (1.1-1.4)1,2 |
| Week 24 | 1.8 (1.6-2.0) | 1.3 (1.1-1.5)1 | 1.2 (1.0-1.4)1 | 1.0 (0.8-1.2)1,2 |
| HAD depression sub-scale, baseline adjusted mean 8.2 | ||||
| Week 8 | 7.0 (6.3-7.6) | 6.1 (5.5-6.7)1 | 5.1 (4.4-5.7)1,2 | 4.8 (4.1-5.4)1,2 |
| Week 24 | 6.5 (5.7-7.2) | 5.6 (4.9-6.3) | 5.0 (4.3-5.8)1 | 3.8 (3.1-4.6)1,2,3 |
| CGI—S, baseline adjusted mean 4.5 | ||||
| Week 8 | 3.3 (3.1-3.5) | 3.1 (2.8-3.3) | 2.8 (2.6-3.0)1 | 2.6 (2.4-2.8)1,2 |
| Week 24 | 3.1 (2.8-3.4) | 2.7 (2.4-2.9)1 | 2.5 (2.3-2.8)1 | 2.2 (1.9-2.4)1,2 |
| BSA total score, baseline adjusted mean 26.0 | ||||
| Week 8 | 17.7 (16.2-19.2) | 15.3 (13.8-16.7)1 | 13.4 (11.9-14.9)1 | 13.0 (11.5-14.5)1,2 |
| Week 24 | 16.9 (15.3-18.6) | 13.8 (12.2-15.4)1 | 11.9 (10.2-13.5)1 | 10.7 (9.1-12.4)1,2 |
| SAS—SR overall, baseline adjusted mean 2.2 | ||||
| Week 8 | 2.0 (1.9-2.1)1,2 | 2.0 (1.9-2.0) | 1.9 (1.8-1.9)1,2 | 1.8 (1.7-1.9)1,2 |
| Week 24 | 1.9 (1.9-2.0) | 1.9 (1.8-2.0) | 1.8 (1.7-1.9)1 | 1.7 (1.7-1.8)1,2 |
Onset of effect
For most variables, onset of anxiolytic effect was seen at weeks 1 and 2 with 150 mg of venlafaxine ER and at weeks 2 and 3 with 75 and 37.5 mg (Table 5). There was a tendency for more parameters to show onset of effect at week 3 with the lowest dose of venlafaxine ER. Onset of effect on the somatic factors of the BSA and the HRSA appeared later in the treatment — at weeks 4 and 8, respectively.
Table 5 Overview of week of onset of efficacy for intention-to-treat sample using the last observation carried forward method
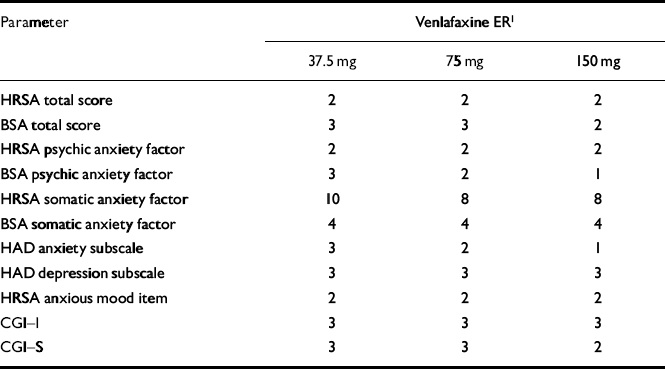
| Parameter | Venlafaxine ER1 | ||
|---|---|---|---|
| 37.5 mg | 75 mg | 150 mg | |
| HRSA total score | 2 | 2 | 2 |
| BSA total score | 3 | 3 | 2 |
| HRSA psychic anxiety factor | 2 | 2 | 2 |
| BSA psychic anxiety factor | 3 | 2 | 1 |
| HRSA somatic anxiety factor | 10 | 8 | 8 |
| BSA somatic anxiety factor | 4 | 4 | 4 |
| HAD anxiety subscale | 3 | 2 | 1 |
| HAD depression subscale | 3 | 3 | 3 |
| HRSA anxious mood item | 2 | 2 | 2 |
| CGI—I | 3 | 3 | 3 |
| CGI—S | 3 | 3 | 2 |
Dose and response
All doses of venlafaxine ER showed significantly higher treatment response rates compared with placebo on both the HRSA and CGI-I as early as week 2. This effect was maintained until week 24 (except for the 37.5 mg venlafaxine ER group at week 8). Responder rates for the CGI-I scores showed a significant difference from placebo and a dose response with respect to onset: 150 mg of venlafaxine ER at week 1, 75 mg of venlafaxine ER at week 2 and 37.5 mg of venlafaxine ER at week 4 (Fig. 2).
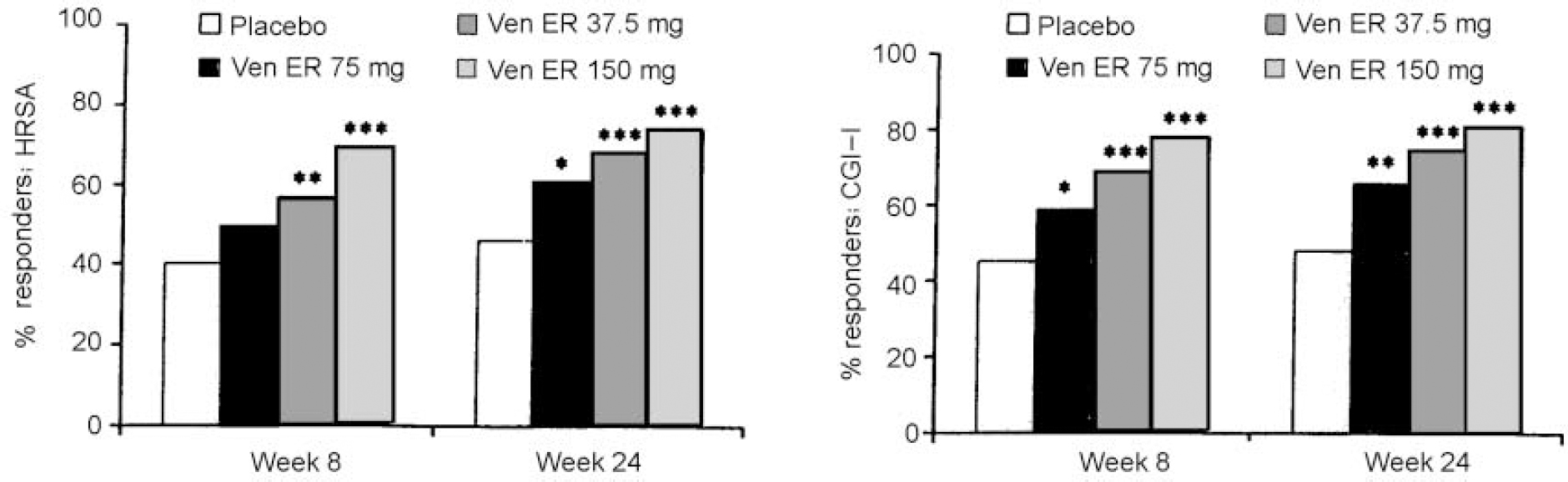
Fig. 2 Percentage of responders at weeks 8 and 24 as assessed by a 50% reduction from baseline score in the Hamilton Rating Scale for Anxiety (HRSA) (left) or a Clinical Global Impression of Improvement (CGI-I) score of 1 or 2 (right): * P<0.05; ** P<0.01 and *** P<0.001 for venlafaxine ER v. placebo in intention-to-treat sample using the last observation carried forward method.
Adverse events
There was no difference with respect to the overall frequency of treatment-emerging adverse events (TEAEs) reported during either short— (82%, 87%, 93%, 88%) or long-term treatment (56%, 62%, 69%, 65%) for the placebo and 37.5-, 75— and 150-mg venlafaxine ER groups, respectively. The most frequent events were nausea, dizziness, sweating, dry mouth and constipation; the incidence of these events appeared to be dose-related and subsided within a short time (Table 6). With respect to sexual dysfunction, the incidence of TEAEs reported on venlafaxine ER was less than 10% at any dose compared with 0% on placebo. The need for concomitant medication or temporary cessation of treatment to manage TEAEs was similar for the placebo and all active treatment groups.
Table 6 Percentage of patients with treatment-emerging adverse events (TEAEs): ≥ 10% incidence and at least twice the incidence seen with placebo

| TEAE | Placebo (n=130) | Venlafaxine ER | ||
|---|---|---|---|---|
| 37.5 mg (n=140) | 75 mg (n=134) | 150 mg (n=137) | ||
| Constipation | 5 | 8 | 13 | 15 |
| Dizziness | 14 | 15 | 22 | 31 |
| Dry mouth | 4 | 6 | 13 | 17 |
| Infection | 4 | 9 | 5 | 12 |
| Nausea | 14 | 22 | 34 | 42 |
| Paraesthesiae | 2 | 1 | 2 | 10 |
| Sweating | 5 | 9 | 11 | 18 |
There were no differences between the treatment groups in the percentage of patients who discontinued and gave adverse events as either a primary or a secondary reason: 22%, 14%, 21% and 19% for placebo, 37.5-, 75— and 150-mg treatment groups, respectively. The most common adverse event leading to discontinuation in the venlafaxine ER treatment groups was nausea: 4%, 6% and 7% for 37.5-, 75— and 150-mg groups, respectively, compared with 2% for placebo. Dizziness was the most frequent cause of discontinuation in the placebo group: 5% compared with <1%, 5% and 4% in the 37.5-, 75— and 150-mg venlafaxine ER treatment groups respectively. Other adverse events associated with discontinuation, but with no apparent association with either placebo or venlafaxine ER, were headache (2% in each group), sweating (<1%, 1%, 4% and 2%) and insomnia (<1%, 1%, 3% and 2%) for placebo and 37.5, 75 and 150 mg of venlafaxine ER respectively. Changes in laboratory parameters, ECG, weight and vital signs, including blood pressure readings, were generally small and sporadic and were not judged to be clinically important.
The potential effects of abrupt discontinuation of venlafaxine ER were evaluated in 83% (297/359) of patients during a 1-week, single-blind placebo discontinuation phase. The proportion of patients reporting at least three new symptoms during the discontinuation phase was similar in the 37.5— and 75-mg venlafaxine ER groups, where the most frequent symptoms were dizziness and/or light— headedness. Discontinuation symptoms with an incidence of ≥10% and double the incidence on placebo were highest with 150 mg of venlafaxine ER, where the most frequent events were dizziness, light— headedness, tinnitus, nausea, vomiting and loss of appetite (Table 7). Discontinuation symptoms began 24-72 h after the last dose of active treatment and usually lasted 3-7 days. There were no differences between treatment groups in the potential for rebound anxiety (defined as a greater HRSA total score during the discontinuation phase than at baseline in patients who had shown a response).
Table 7 Percentage of patients with discontinuation symptoms on the Benzodiazepine Withdrawal Symptom Questionnaire (Reference Tyrer, Murphy and RileyTyrer et al, 1990): ≥ 10% incidence and at least twice the incidence seen with placebo

| Symptoms | Placebo (n=71) | Venlafaxine ER | ||
|---|---|---|---|---|
| 37.5 mg (n=82) | 75 mg (n=80) | 150 mg (n=84) | ||
| Dizziness | < 1 | 15 | 10 | 13 |
| Light-headedness | 4 | 15 | 8 | 12 |
| Tinnitus | < 1 | 2 | 4 | 13 |
| Nausea | 3 | 9 | 6 | 18 |
| Vomiting | < 1 | 5 | 3 | 12 |
| Loss of appetite | 3 | 6 | 4 | 10 |
DISCUSSION
Although immediate relief of morbid anxiety can be obtained with the benzodiazepines and sometimes with beta-blockers, the only available option for maintenance treatment of chronic GAD has been buspirone, a compound with lower comparative efficacy and more frequent treatment interruptions owing to side-effects (Reference Schweizer, Rickels and LuckiSchweizer et al, 1986). There is, however, no placebo-controlled evidence of buspirone in long-term therapy. Other anxiety disorders, such as panic disorder, obsessive-compulsive disorder and post-traumatic stress disorder, are all responsive to treatment with serotonergic reuptake inhibitors (Reference Zohar and WestenbergZohar & Westenberg, 2000).
Efficacy
Efficacy for the two higher doses of venlafaxine ER (75 and 150 mg daily) was evident across all the primary comparisons. These effects were maintained during treatment for up to 6 months. Results for 37.5 mg venlafaxine ER were not as robust or consistent. Corroborating evidence for efficacy is found in the results of the secondary efficacy measures, including improvement of impairment in social functioning. The improved social functioning associated with treatment with venlafaxine ER will be reported in detail in a separate paper.
These assessments of efficacy indicated a dose-response relationship, with the highest dose of venlafaxine ER (150 mg) showing the greatest improvements and highest responder rates. This was significant compared with the lowest dose (37.5 mg) at weeks 8 and 24. The dose-response relationship was also apparent for onset of the anxiolytic effect. Onset was seen from weeks 1 or 2 with 150 mg of venlafaxine ER, from weeks 2 or 3 with the 75-mg dose and from weeks 2-4 with the 37.5-mg dose for all variables apart from somatic anxiety. The delay in improvement of somatic anxiety symptoms may reflect the natural course of improvement in anxiety, the side-effects seen during the first 2 weeks of venlafaxine ER treatment or the differences in response to either venlafaxine Er or placebo for psychic and somatic anxiety.
Tolerance
In any chronic condition where long-term treatment is the norm, it is important that the intervention is not only safe and well tolerated but also that there is good patient acceptability. The similar overall discontinuation rates for all treatment groups, including placebo, and the similar discontinuation rates where adverse events were cited either as a primary or secondary reason suggest good patient acceptability of venlafaxine ER in the management of GAD. The benign safety profile (laboratory, blood pressure, weight and ECG variables) of venlafaxine ER in the dose range up to 150 mg daily also was apparent in this population.
Discontinuation
The experimental design employed here included an evaluation of the extent of discontinuation symptoms following abrupt discontinuation of all three fixed doses of venlafaxine ER. The findings of dose-related symptoms during the discontinuation phase are consistent with the current understanding and experience with venlafaxine and the wording of the labelling for the depression indication, where it is recommended that doses above 75 mg of venlafaxine ER should be tapered before discontinuation.
Similar recommendations are valid for all of the selective serotonin reuptake inhibitors. Importantly, there was no evidence for the occurrence of rebound anxiety with any of the doses of venlafaxine ER when treatment was discontinued, as is the case with benzodiazepines (Reference Rickels, Schweizer and CaseRickels et al, 1990). Physical discontinuation symptoms are known to be associated with a number of commonly used psychoactive compounds, including the serotonin reuptake inhibitors (Reference Rosenbaum, Fava and HoogRosenbaum et al, 1998).
Dose
The current study provides evidence for the efficacy of venlafaxine ER in both the short— and long-term treatment of GAD and the efficacy is dose-related over the range studied. The optimal clinical dose of venlafaxine ER is 75 mg daily in most cases requiring the management of symptoms of anxiety. In some patients, and when clinically indicated, it may be necessary to increase the dose of venlafaxine ER to 150 mg daily.
Venlafaxine in vitro inhibits the reuptake of both serotonin and noradrenaline, although the relative potencies at the sites and the interpretation of the clinical meaning of these findings have been discussed. It has been suggested that noradrenaline effects in humans only become apparent at higher doses. In recent studies, however, enhancement of noradrenaline activity was found at either 75 or 150 mg (Reference Abdelmawal, Langley and BradshawAbdelmawal et al, 1999, Reference Bitsios, Szabadi and BradshawBitsios et al, 1999). Melichar et al (Reference Melichar, Haida and Rhodes2001) demonstrated that 1 mg/kg venlafaxine displaced the noradrenaline ligand MHED. These data suggest that venlafaxine inhibits the reuptake of both monoamines at the lower end of the dose range, but that the full effect on noradrenaline may require 150 mg or more.
Bearing in mind the chronicity of GAD and the frequent likely comorbidity with other Axis I disorders, further studies extending beyond 6 months evaluating the effect of venlafaxine ER and studies in comorbid populations are also recommended.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
• Seventy per cent of patients with generalised anxiety disorder responded to treatment with venlafaxine extended release (ER) at 75 or 150 mg/day.
-
• The efficacy of venlafaxine ER was maintained over six months and the treatment interruption rate was low.
-
• Discontinuation symptoms after higher doses call for dose tapering.
LIMITATIONS
-
• The prevention of relapse in responders to venlafaxine ER was not assessed.
-
• Potential age and gender differences in response were not evaluated.
-
• The design did not study the optimal dose for individual patients.
APPENDIX
List of investigators
Belgium: M. Dierick, Psychiatrist, St Denijs-Westrem; P. Elens, Neuropyschiatrist, Munsterbilzen; A. Mertens, Psychiatrist, St Denijs-Westrem; B. Mertens, Psychiatrist, St Denijs-Westrem; R. Spiers, Neuro-psychiatrist, St Denijs-Westrem. Finland: H. Jokela, Chief Physician of Psychiatric Department, Lahti; J. Kärkkäinen, Psychiatrist, Pori; H. Pekkarinen, Senior Lecturer in Psychiatry, Tampere; M. Pirilä, Senior Consultant in Psychiatry, Tampere; A. Torppa, General Practitioner, Helsinki. France: R. Arnaud-Castiglioni, Hospital Psychiatrist, Marseille; R. Azra, Psychiatrist in Private Practice, Strasbourg; A. Bertolino, General Practitioner, Marseille; X. Brunier, General Practitioner, Pont St Martin; E. Cortell, Private Practice, Marseille; B. Cyran, Hospital Psychiatrist, Lens; A. Deroche, Psychiatrist in Private Practice, Joue-Les-Tours; J. M. Ehrlich, General Practitioner, Strasbourg; J. J. Elkouri, General Practitioner in Private Practice, Nantes; B. Ermolenko, General Practitioner in Private Practice, Lambersat; D. Hindennach, General Practitioner, Marseille; E. Hirsch, General Practitioner in Private Practice, Strasbourg; J. F. Katz, Psychiatrist in Private Practice, Saint-Brieuc; C. G. Lafont, General Practitioner, Marseille; R. Laroche, General Practitioner, Marseille; G. Leibovici, Psychiatrist, Marseille; J. M. Letzelter, General Practitioner, Strasbourg; P. Marmor, General Practitioner in Private Practice, Strasbourg; P. Platel, General Practitioner, Lille; Y. L. Raoul, Teacher in Psychiatry, Toulon Naval; M. H. Vanderpotte, General Practitioner, Faches Thumesnil; A. Wurtz, General Practitioner, Strasbourg; J. R. Zekri, Psychiatrist in Private Practice, Marseille; M. Zins-Ritter, Psychiatrist, Nantes. Sweden: C. Allgulander, Associate Professor of Psychiatry, Karolinska Institutet; I. Sjödin, Associate Professor in Psychiatry, Linköping University. UK: M. Adler, Principal in General Practice, Harrow; R. Cook, General Practice, Saltash; A. Cowie, Principal in General Practice, Corsham; N. Gray, General Practitioner, Redruth; W. Jago, Principal in General Practice, Penzance; I. G. James, Principal in General Practice, Bolton; W. Ahmed, Consultant in Psychiatry, Taunton; P. Barton, General Practitioner, Helston; M. Blagden, General










eLetters
No eLetters have been published for this article.