


Psychological reactions to traumatic events such as post-traumatic stress disorder (PTSD) are common and clinically important, with PTSD shown to have a lifetime prevalence in community samples of between 1.3% (Reference Helzer, Robins and McEvoyHelzer et al, 1987) and 9% (Reference Breslau, Davis and AndreskiBreslau et al, 1991). Research has confirmed the complex aetiology, epidemiology and symptomatology of PTSD and demonstrated a wide range of neurobiological changes (Reference FriedmanFriedman, 1997). Neuroimaging techniques provide opportunities to investigate structural and functional brain abnormalities, with the challenge then to integrate ever more complex neurobiological models across many neurochemical systems and structures into a cohesive understanding of PTSD.
This review examines structural and functional studies of the brain and also studies combining both modalities. Symptom and non-symptom provocation paradigms are examined in relation to functional studies, with case reports reviewed separately. Finally, the implications of these findings are discussed.
METHOD
Neuroimaging studies of subjects with PTSD were identified using literature searches of the Medline, Psychlit, Embase and Published International Literature on Traumatic Stress (PILOTS) databases, current contents, recent journal issues, reference lists and secondary sources (e.g. book chapters). A total of 30 published reports were located: 12 reports of brain structure in PTSD and 18 reports of brain function in PTSD (with two of these studies combining the imaging modalities). All English language articles published by August 2001 were included in this review.
RESULTS
Studies of brain structure in patients with PTSD
Computed tomography studies were first to suggest brain abnormalities in PTSD (Table 1) and there have been eight reports on magnetic resonance imaging (MRI) studies in subjects with PTSD (Table 2).
Table 1 Computed tomography studies of patients with post-traumatic stress disorder (PTSD)

| Author | Subjects | Control/comparison groups | Diagnostic criteria (method) | Results |
|---|---|---|---|---|
| Reference Silverman, Hart and StockmanSilverman et al, 1989 | 14 exposed to neurotoxin (pentaborane); 7 with PTSD (7M; 7F) | None | DSM—III (Diagnostic Interview Schedule) | Abnormal VBRs at 1-3 and 18 months post-exposure; no association with PTSD or other diagnosis; abnormal VBRs may be due to neurotoxin |
| Reference Peters, van Kammen and van KammenPeters et al, 1990 | 10 former prisoners of war (all M) | None | Not specified | Increased VBR and sulcal widening associated with sleep disturbances |
Table 2 Magnetic resonance imaging studies of patients with post-traumatic stress disorder (PTSD)
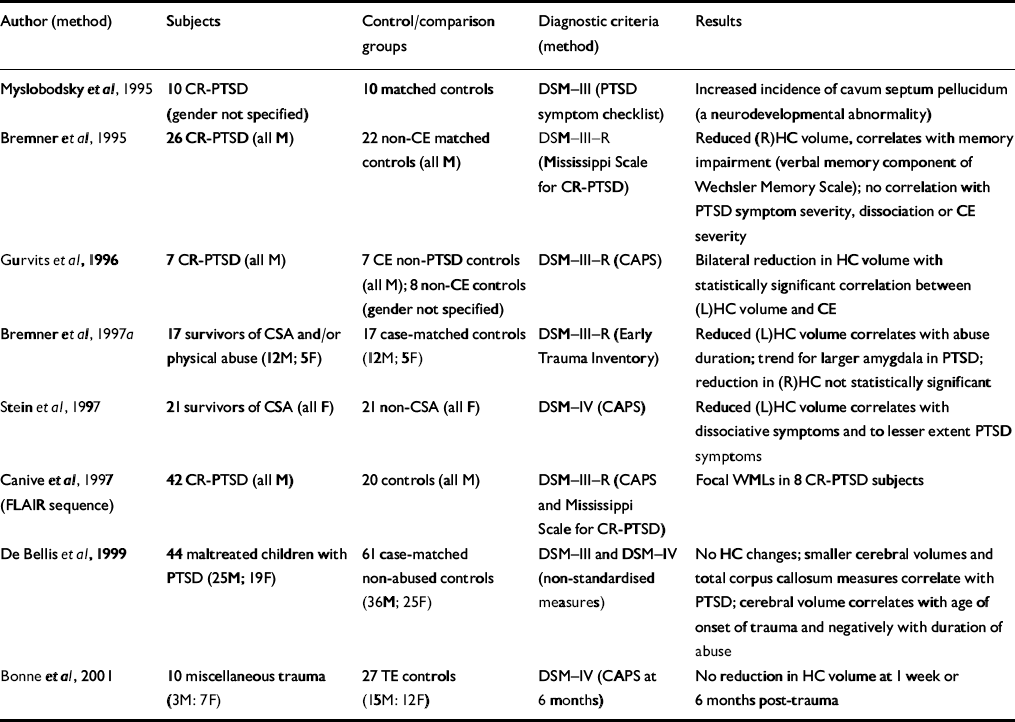
| Author (method) | Subjects | Control/comparison groups | Diagnostic criteria (method) | Results |
|---|---|---|---|---|
| Reference Myslobodsky, Glicksohn and SingerMyslobodsky et al, 1995 | 10 CR-PTSD (gender not specified) | 10 matched controls | DSM—III (PTSD symptom checklist) | Increased incidence of cavum septum pellucidum (a neurodevelopmental abnormality) |
| Reference Bremner, Randall and ScottBremner et al, 1995 | 26 CR-PTSD (all M) | 22 non-CE matched controls (all M) | DSM—III—R (Mississippi Scale for CR-PTSD) | Reduced (R)HC volume, correlates with memory impairment (verbal memory component of Wechsler Memory Scale); no correlation with PTSD symptom severity, dissociation or CE severity |
| Reference Gurvits, Shenton and HokamaGurvits et al, 1996 | 7 CR-PTSD (all M) | 7 CE non-PTSD controls (all M); 8 non-CE controls (gender not specified) | DSM—III—R (CAPS) | Bilateral reduction in HC volume with statistically significant correlation between (L)HC volume and CE |
| Reference Bremner, Randall and VermettenBremner et al, 1997a | 17 survivors of CSA and/or physical abuse (12M; 5F) | 17 case-matched controls (12M; 5F) | DSM—III—R (Early Trauma Inventory) | Reduced (L)HC volume correlates with abuse duration; trend for larger amygdala in PTSD; reduction in (R)HC not statistically significant |
| Reference Stein, Koverola and HannaStein et al, 1997 | 21 survivors of CSA (all F) | 21 non-CSA (all F) | DSM—IV (CAPS) | Reduced (L)HC volume correlates with dissociative symptoms and to lesser extent PTSD symptoms |
| Reference Canive, Lewine and OrrisonCanive et al, 1997 (FLAIR sequence) | 42 CR-PTSD (all M) | 20 controls (all M) | DSM—III—R (CAPS and Mississippi Scale for CR-PTSD) | Focal WMLs in 8 CR-PTSD subjects |
| Reference De Bellis, Keshavan and ClarkDe Bellis et al, 1999 | 44 maltreated children with PTSD (25M; 19F) | 61 case-matched non-abused controls (36M; 25F) | DSM—III and DSM—IV (non-standardised measures) | No HC changes; smaller cerebral volumes and total corpus callosum measures correlate with PTSD; cerebral volume correlates with age of onset of trauma and negatively with duration of abuse |
| Reference Bonne, Brandes and GilboaBonne et al, 2001 | 10 miscellaneous trauma (3M: 7F) | 27 TE controls (15M: 12F) | DSM—IV (CAPS at 6 months) | No reduction in HC volume at 1 week or 6 months post-trauma |
A number of studies have now demonstrated the involvement of the hippocampus in chronic PTSD. Two studies have examined the hippocampus without finding volumetric changes. The first study demonstrated the absence of hippocampal atrophy prior to the onset of PTSD (Reference Bonne, Brandes and GilboaBonne et al, 2001). Further, after single-event trauma, hippocampal abnormalities did not occur within 6 months. The second study (Reference De Bellis, Keshavan and ClarkDe Bellis et al, 1999) demonstrated smaller cerebral volumes and corpus callosum measures but no hippocampal changes in a group of maltreated children.
Possible reasons for the differences in the extent of volume reduction may be the study design (thickness of slices or whether or not the whole brain volume was calculated), the control group utilised, comorbid substance misuse, treatment effects or neurodevelopmental plasticity. In the main, researchers have performed comparative analyses to minimise the effect of other psychiatric disorders, including alcohol and substance misuse.
Sapolsky (Reference Sapolsky1992) has described the sensitivity of hippocampal formation to severe, chronic traumatic stress and perhaps also to elevated glucocorticoids and/or excitatory amino acids, yet many other disorders such as bipolar disorder, schizophrenia, alcohol misuse and dementia are also associated with hippocampal atrophy (Reference McEwenMcEwen, 1997). Glucocorticoid-induced atrophy would appear to require prolonged or repeated bursts of glucocorticoid excess. However, basal glucocorticoid levels have been shown to be lower than normal in PTSD, perhaps due to enhanced adrenocortical sensitivity to feedback regulation (Yehudaet al, Reference Yehuda, Southwick and Nussbaum1991, Reference Yehuda, Southwick and Krystal1993).
The lateralisation of hippocampal damage has varied, with one possible explanation being changing vulnerability of the hippocampus to stress-induced damage at different developmental stages. The possibility that small hippocampi represent a predisposition to PTSD is not supported by the one study to examine acute PTSD (Reference Bonne, Brandes and GilboaBonne et al, 2001) because no change in volume was present acutely or within the first 6 months. Further, Bremner et al (Reference Bremner, Randall and Scott1995) demonstrated no volume difference between early-onset (before age 8 years) and late-onset (age 8 years or later) abuse.
Canive et al (Reference Canive, Lewine and Orrison1997) have uniquely demonstrated focal white matter lesions (WMLs) in eight subjects within a sample of 42 male combat-exposed subjects. Most of the WMLs were identified using the fluid attenuated inversion recovery (FLAIR) imaging sequence, which is not employed in typical MRI studies. The FLAIR sequence causes suppression of the cerebrospinal fluid signal with WML remaining bright, and the absence of the FLAIR imaging sequence may explain why other studies have not replicated this finding.
Studies using proton magnetic resonance spectroscopy in patients with PTSD
Proton magnetic resonance spectroscopy (MRS) can provide information about alterations in N-acetyl aspartate (NAA) and choline-containing compounds in the human brain without the radiation exposure of positron emission tomography (PET) on single photon emission computed tomography (SPECT) but with lower sensitivity. Two studies have utilised this technique (Table 3) to measure NAA in subjects with PTSD. Schuff et al (Reference Schuff, Marmar and Weiss1997) utilised both MRI and proton MRS to measure hippocampal volume and changes in NAA. An 18% reduction in right hippocampal NAA compared with a 6% reduction in right hippocampal volume suggests that NAA is a more sensitive measure of neuronal loss than volume changes. Although this study does not answer whether the NAA changes were pre-existing or a consequence of trauma rather than PTSD per se, the second study (Reference Freeman, Cardwell and KarsonFreeman et al, 1998) suggests that it correlated with PTSD. Decreased NAA has been demonstrated in other disorders, such as early-onset schizophrenia (Reference Bertolino, Nawroz and MattayBertolino et al, 1996), temporal lobe epilepsy (Reference Ende, Laxer and KnowltonEnde et al, 1997) and Alzheimer's disease (Reference Mackay, Ezekiel and SelafaniMacKay et al, 1996).
Table 3 Functional magnetic resonance imaging (MRI) and magnetic resonance spectroscopy (MRS) studies in patients with post-traumatic stress disorder (PTSD)
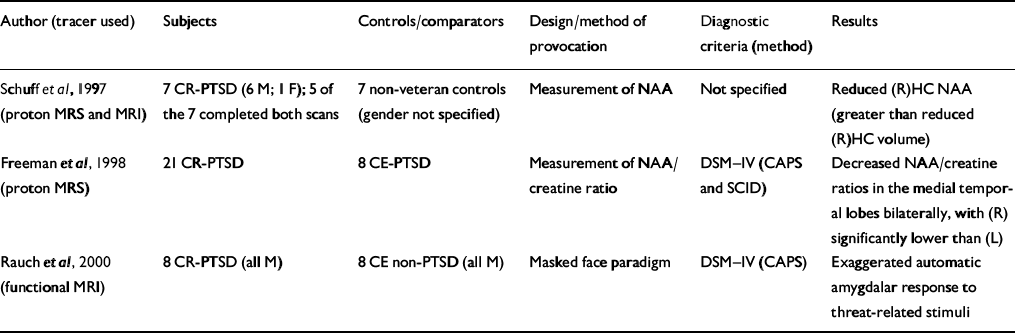
| Author (tracer used) | Subjects | Controls/comparators | Design/method of provocation | Diagnostic criteria (method) | Results |
|---|---|---|---|---|---|
| Reference Schuff, Marmar and WeissSchuff et al, 1997 (proton MRS and MRI) | 7 CR-PTSD (6 M; 1 F); 5 of the 7 completed both scans | 7 non-veteran controls (gender not specified) | Measurement of NAA | Not specified | Reduced (R)HC NAA (greater than reduced (R)HC volume) |
| Reference Freeman, Cardwell and KarsonFreeman et al, 1998 (proton MRS) | 21 CR-PTSD | 8 CE-PTSD | Measurement of NAA/creatine ratio | DSM—IV (CAPS and SCID) | Decreased NAA/creatine ratios in the medial temporal lobes bilaterally, with (R) significantly lower than (L) |
| Reference Rauch, Whalen and ShinRauch et al, 2000 (functional MRI) | 8 CR-PTSD (all M) | 8 CE non-PTSD (all M) | Masked face paradigm | DSM—IV (CAPS) | Exaggerated automatic amygdalar response to threat-related stimuli |
Studies of brain function in patients with PTSD
Most functional brain imaging studies in PTSD have used either PET or SPECT, which involve the detection of radiation-emitting radioisotopes to measure regional cerebral metabolism or blood flow. Functional MRI determines regional brain activation by detecting changes in blood oxygenation level and has a better spatial resolution than PET or SPECT.
The majority of studies employed a symptom provocation paradigm because measuring brain function at rest poses the problem of controlling the range of possible mental states. Studies have concentrated upon traumatic memory as a central component in PTSD because the presence of intrusive symptoms compellingly points to PTSD as the diagnosis. The one study to employ a non-symptom provocation paradigm (Reference Lucey, Costa and AdsheadLuceyet al, 1997) used SPECT to study three groups with anxiety-related disorders: PTSD, obsessive—compulsive disorder (OCD) and agoraphobia with panic disorder (see Table 5). One study utilised a pharmacological challenge and is discussed separately below (Reference Bremner, Innis and NgBremner et al, 1997b ).
Table 4 Positron emission tomography (PET) studies of patients with post-traumatic stress disorder (PTSD)
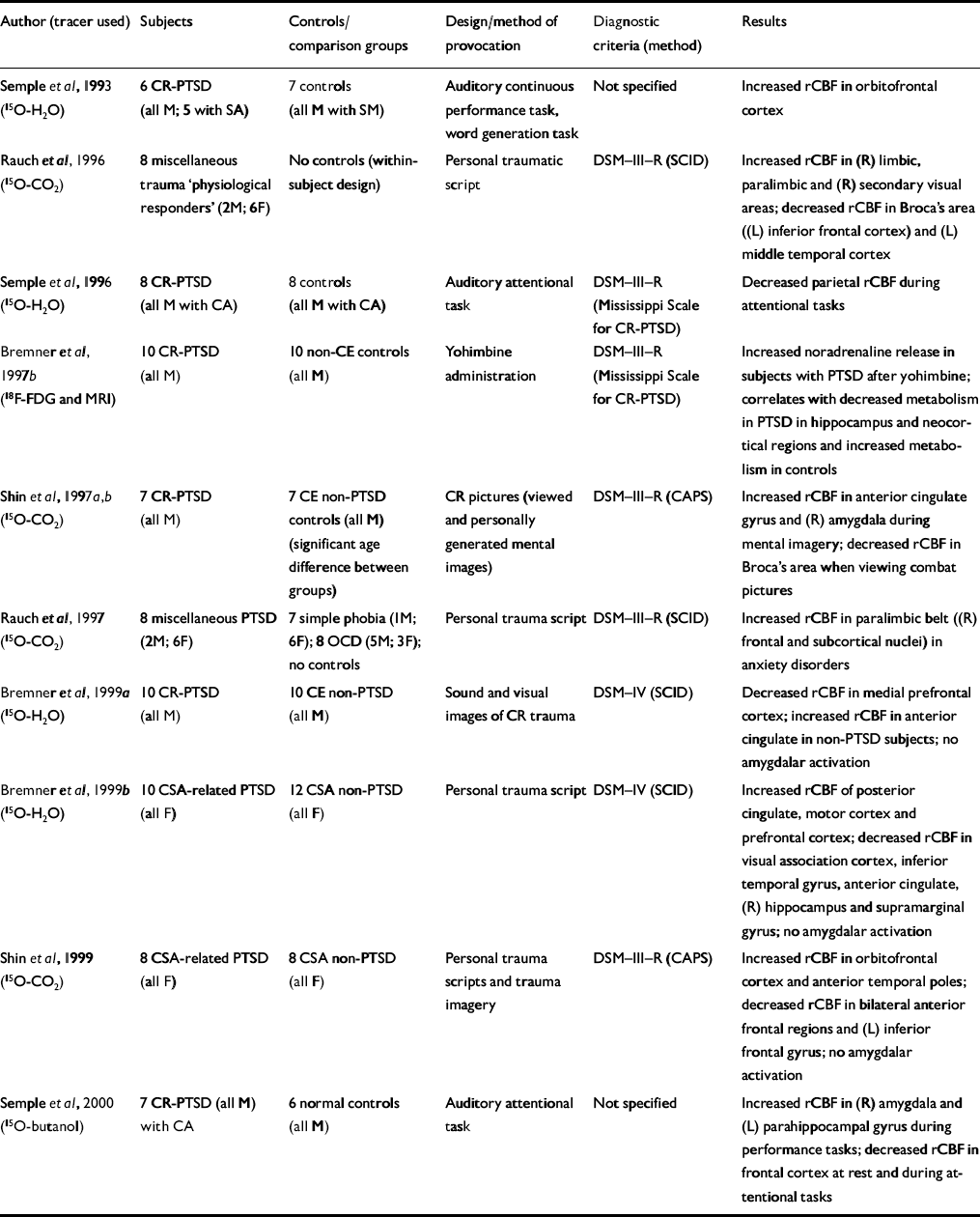
| Author (tracer used) | Subjects | Controls/comparison groups | Design/method of provocation | Diagnostic criteria (method) | Results |
|---|---|---|---|---|---|
| Reference Semple, Goyer and McCormickSemple et al, 1993 (15O-H2O) | 6 CR-PTSD (all M; 5 with SA) | 7 controls (all M with SM) | Auditory continuous performance task, word generation task | Not specified | Increased rCBF in orbitofrontal cortex |
| Reference Rauch, van Der Kolk and FisherRauch et al, 1996 (15O-CO2) | 8 miscellaneous trauma ‘physiological responders’ (2M; 6F) | No controls (within-subject design) | Personal traumatic script | DSM—III—R (SCID) | Increased rCBF in (R) limbic, paralimbic and (R) secondary visual areas; decreased rCBF in Broca's area ((L) inferior frontal cortex) and (L) middle temporal cortex |
| Reference Semple, Goyer and McCormickSemple et al, 1996 (15O-H2O) | 8 CR-PTSD (all M with CA) | 8 controls (all M with CA) | Auditory attentional task | DSM—III—R (Mississippi Scale for CR-PTSD) | Decreased parietal rCBF during attentional tasks |
| Reference Bremner, Innis and NgBremner et al, 1997b (18F-FDG and MRI) | 10 CR-PTSD (all M) | 10 non-CE controls (all M) | Yohimbine administration | DSM—III—R (Mississippi Scale for CR-PTSD) | Increased noradrenaline release in subjects with PTSD after yohimbine; correlates with decreased metabolism in PTSD in hippocampus and neocortical regions and increased metabolism in controls |
| Shin et al, Reference Shin, Kosslyn and McNally1997a ,Reference Shin, McNally and Kosslyn b (15O-CO2) | 7 CR-PTSD (all M) | 7 CE non-PTSD controls (all M) (significant age difference between groups) | CR pictures (viewed and personally generated mental images) | DSM—III—R (CAPS) | Increased rCBF in anterior cingulate gyrus and (R) amygdala during mental imagery; decreased rCBF in Broca's area when viewing combat pictures |
| Reference Rauch, Savage and AlpertRauch et al, 1997 (15O-CO2) | 8 miscellaneous PTSD (2M; 6F) | 7 simple phobia (IM; 6F); 8 OCD (5M; 3F); no controls | Personal trauma script | DSM—III—R (SCID) | Increased rCBF in paralimbic belt ((R) frontal and subcortical nuclei) in anxiety disorders |
| Reference Bremner, Staib and KaloupekBremner et al, 1999a (15O-H2O) | 10 CR-PTSD (all M) | 10 CE non-PTSD (all M) | Sound and visual images of CR trauma | DSM—IV (SCID) | Decreased rCBF in medial prefrontal cortex; increased rCBF in anterior cingulate in non-PTSD subjects; no amygdalar activation |
| Reference Bremner, Narayan and StaibBremner et al, 1999b (15O-H2O) | 10 CSA-related PTSD (all F) | 12 CSA non-PTSD (all F) | Personal trauma script | DSM—IV (SCID) | Increased rCBF of posterior cingulate, motor cortex and prefrontal cortex; decreased rCBF in visual association cortex, inferior temporal gyrus, anterior cingulate, (R) hippocampus and supramarginal gyrus; no amygdalar activation |
| Reference Shin, McNally and KosslynShin et al, 1999 (15O-CO2) | 8 CSA-related PTSD (all F) | 8 CSA non-PTSD (all F) | Personal trauma scripts and trauma imagery | DSM—III—R (CAPS) | Increased rCBF in orbitofrontal cortex and anterior temporal poles; decreased rCBF in bilateral anterior frontal regions and (L) inferior frontal gyrus; no amygdalar activation |
| Reference Semple, Goyer and McCormickSemple et al, 2000 (15O-butanol) | 7 CR-PTSD (all M) with CA | 6 normal controls (all M) | Auditory attentional task | Not specified | Increased rCBF in (R) amygdala and (L) parahippocampal gyrus during performance tasks; decreased rCBF in frontal cortex at rest and during attentional tasks |
Table 5 Single photon emission computed tomography (SPECT) studies in patients with post-traumatic stress disorder (PTSD)
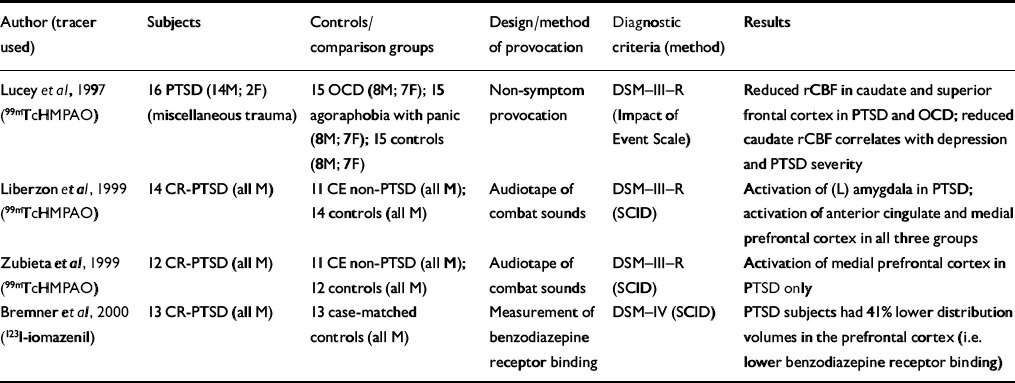
| Author (tracer used) | Subjects | Controls/comparison groups | Design/method of provocation | Diagnostic criteria (method) | Results |
|---|---|---|---|---|---|
| Reference Lucey, Costa and AdsheadLucey et al, 1997 (99mTcHMPAO) | 16 PTSD (14M; 2F) (miscellaneous trauma) | 15 OCD (8M; 7F); 15 agoraphobia with panic (8M; 7F); 15 controls (8M; 7F) | Non-symptom provocation | DSM—III—R (Impact of Event Scale) | Reduced rCBF in caudate and superior frontal cortex in PTSD and OCD; reduced caudate rCBF correlates with depression and PTSD severity |
| Reference Liberzon, Taylor and AmdurLiberzon et al, 1999 (99mTcHMPAO) | 14 CR-PTSD (all M) | 11 CE non-PTSD (all M); 14 controls (all M) | Audiotape of combat sounds | DSM—III—R (SCID) | Activation of (L) amygdala in PTSD; activation of anterior cingulate and medial prefrontal cortex in all three groups |
| Reference Zubieta, Chinitz and LombardiZubieta et al, 1999 (99mTcHMPAO) | 12 CR-PTSD (all M) | 11 CE non-PTSD (all M); 12 controls (all M) | Audiotape of combat sounds | DSM—III—R (SCID) | Activation of medial prefrontal cortex in PTSD only |
| Reference Bremner, Innis and SouthwickBremner et al, 2000 (123l-iomazenil) | 13 CR-PTSD (all M) | 13 case-matched controls (all M) | Measurement of benzodiazepine receptor binding | DSM—IV (SCID) | PTSD subjects had 41% lower distribution volumes in the prefrontal cortex (i.e. lower benzodiazepine receptor binding) |
Pharmacological challenge study
Bremner et al (Reference Bremner, Innis and Ng1997b ) administered the α2-antagonist yohimbine in a pharmacological challenge symptom provocation paradigm (Table 4). Yohimbine has been shown to provoke an exaggerated behavioural and biochemical responsiveness in subjects with PTSD (Reference Southwick, Krystal and MorganSouthwick et al, 1993) and panic disorder (Reference Charney, Woods and GoodmanCharney et al, 1987a ) but not in patients with other mental disorders (Reference Charney, Woods and HeningerCharney et al, 1987b ; Reference Glazer, Charney and HeningerGlazer et al, 1987; Reference Rasmussen, Goodman and WoodsRasmussenet al, 1987; Reference Heninger, Charney and PriceHeningeret al, 1988). Bremner et al demonstrated that yohimbine administration correlated with increased anxiety symptoms in patients with PTSD but not in controls. Patients with yohimbine-induced panic attack (approximately 60%) had significantly reduced hippocampal and neocortical metabolism, suggesting enhanced noradrenaline release in subjects with PTSD after yohimbine administration.
Symptom provocation studies using PET
The majority of PET studies (six reports) provoked intrusive symptoms by using personalised trauma scripts or trauma-related sounds or pictures (Table 4), and one group examined changes during the performance of cognitive tasks (Semple et al, Reference Semple, Goyer and McCormick1993, Reference Semple, Goyer and McCormick1996, Reference Semple, Goyer and McCormick2000).
Symptom provocation studies using SPECT
Only three SPECT studies have examined subjects with PTSD after symptom provocation (Table 5). Liberzonet al have conducted a SPECT study of subjects with combat-related PTSD and reported key findings in two papers (Reference Liberzon, Taylor and AmdurLiberzon et al, 1999; Reference Zubieta, Chinitz and LombardiZubieta et al, 1999) and one case report (Reference Liberzon, Taylor and FigLiberzon et al, 1996/97). They report separately on a specific analysis of the activity of the medial prefrontal cortex, which is thought to modulate the fear response (Reference Zubieta, Chinitz and LombardiZubieta et al, 1999). An increased regional cerebral blood flow (rCBF) was found in the medial prefrontal cortex in subjects with PTSD during provocation, although no statistically significant association was found with peripheral measures. The different findings may reflect the fact that the subjects were from the same cohort but the region-of-interest analysis was derived from a different cohort of normal controls. The anatomical region sampled for the region-of-interest analysis was more rostral than the anterior cingulate region examined by Liberzon et al (Reference Liberzon, Taylor and Amdur1999).
Functional MRI studies
Rauch et al (Reference Rauch, Whalen and Shin2000) conducted a functional MRI study applying a validated method for measuring automatic amygdala responses to general threat-related stimuli using a masked face paradigm (Reference Whalen, Rauch and EtcoffWhalen et al, 1998b ). The magnitude of amygdala response distinguished subjects with PTSD from those without PTSD with 75% sensitivity and 100% specificity (Table 3).
Case reports involving functional neuroimaging
Levin et al (Reference Levin, Lazrove and van der Kolk1999) report on one subject from an ongoing SPECT study utilising the same provocation paradigm as Rauch et al (Reference Rauch, van Der Kolk and Fisher1996). Subjects were scanned before and after successful treatment for PTSD. After treatment there were two areas of increased activity: the anterior cingulate cortex and the left frontal lobe. The treatment was eye movement desensitisation and reprocessing (EMDR; Reference ShapiroShapiro, 1996) but the subject of this report was also on a selective serotonin reuptake inhibitor throughout the study. The authors state that these changes were consistent with summed data from four out of six subjects in their ongoing study.
Profound memory deficits were demonstrated in a case report of a man after a second traumatic event triggering memories of a fatal accident he witnessed as a child. A PET scan revealed hypoperfusion in memory-sensitive regions such as the hippocampal formation and temporal lobe (Reference Markowitsch, Kessler and Van Der VenMarkowitsch et al, 1998). Liberzon et al (Reference Liberzon, Taylor and Fig1996/97) report one patient who experienced a flashback after provocation while being scanned. There was greater uptake in subcortical regions, especially the thalamus, in comparison with cortical regions.
Summary of functional findings
Many of the functional neuroimaging studies have suggested abnormalities of limbic and paralimbic areas during symptom provocation and cognitive activation studies. This supports the suggested role of these areas in mediating emotional arousal in normal anxiety (Reference Benkelfat, Bradwejn and MeyerBenkelfat et al, 1996), across a range of disorders (Reference Rauch, Savage and AlpertRauch et al, 1995) and in trauma-exposed non-PTSD groups. These changes are therefore not specific to PTSD. Hyperperfusion of limbic and paralimbic areas may be a result of stress-induced long-term potentiation of the monosynapticN-methyl-D-aspartate (NMDA)-mediated pathway between the amygdala and the periacqueductal grey (Reference Davidson and SuttonDavidson & Sutton, 1995; Reference AdamecAdamec, 1997). The NMDA receptors are thought to be activated to produce long-term memories of events when sufficient glutamate is released as a result of the stress (Reference Glue, Nutt, Coupland, Stanford and SalmonGlue et al, 1993).
Amygdalar activation may be detected more easily during the processing of fear-related stimuli (Reference Breiter, Etcoff and WhalenBreiter et al, 1996; Reference Morris, Frith and PerrettMorris et al, 1996; Reference Whalen, Bush and McNallyWhalen et al, 1998a ). Inconsistent findings of amygdala activation may reflect the nature of the trauma, and the greater emotional responsiveness to personal narratives of traumatic events (Reference Rauch, van Der Kolk and FisherRauch et al, 1996) rather than to generalised trauma-related pictures and images (Shin et al, Reference Shin, Kosslyn and McNally1997a ,Reference Shin, McNally and Kosslyn b ) or to generalised combat sounds (Reference Bremner, Staib and KaloupekBremneret al, 1999a ). For example, Shin et al (Reference Shin, McNally and Kosslyn1997b ) found increased activation of the amygdala in PTSD only during combat imagery, despite both the imagery and combat perception conditions being rated as of equal significance and causing equal arousal. In addition, the failure to demonstrate amygdalar activation may be due to its involvement in encoding an event's emotional significance but not in the recall of the event per se (Reference Cahill, Haier and FallonCahill et al, 1996).
The role of the amygdala was investigated in subjects with probable Alzheimer's disease who experienced the 1995 earthquake in Kobe, Japan (Reference Ikeda, Mori and HironoIkeda et al, 1998; Reference Mori, Ikeda and HironoMori et al, 1999; Reference Kazui, Mori and HashimotoKazui et al, 2000). Memories of the earthquake were examined as an index of emotional memory. Subjects remembered their personal experience rather than the context of the earthquake, and emotional memory correlated with normalised amygdalar volume (via MRI) irrespective of generalised brain atrophy and cognitive impairments.
Increased automatic amygdalar responsiveness to stimuli (cognitive activation or masked face paradigm) has been shown to be accompanied by decreased activity of the prefrontal cortex, which has a role in the encoding and retrieval of verbal memories. Lower benzodiazepine receptor binding in the prefrontal cortex might mean that PTSD causes a down-regulation of benzodiazepine receptor binding or that pre-trauma low levels of benzodiazepine receptor binding in the prefrontal cortex might increase the risk of developing PTSD after traumatic events (Reference Bremner, Innis and SouthwickBremner et al, 2000).
Although needing replication, the study by Levin et al (Reference Levin, Lazrove and van der Kolk1999) suggests that successful treatment may not only reduce amygdala activity but also may involve activation of structures implicated in the modulation of fight/flight reactions to perceived threat, and perhaps the differentiation of real from imagined threat. Increased perfusion of the thalamus during a flashback (Reference Liberzon, Taylor and FigLiberzon et al, 1996/97) supports its suggested role in the generation of dissociative symptoms in PTSD (Reference Krystal, Bennett, Bremner, Friedman, Charney and DeutchKrystalet al, 1995).
The absence of increased anterior cingulate activation over comparison groups may be associated with the inability of people with PTSD to extinguish fear. It is thought to play a major role in the assignment of motivational significance and is associated with non-specific anxiety states being activated in procaine-induced fear (Reference Ketter, Andreason and GeorgeKetteret al, 1996), imagery of aversive stimuli (Reference Kosslyn, Shin and ThompsonKosslyn et al, 1996) and healthy individuals recollecting sad events (Reference Whalen, Bush and McNallyWhalen et al, 1998a ). The increased activation of the posterior cingulate (Reference Bremner, Narayan and StaibBremner et al, 1999b ) may relate to its suggested role in the emotional processing of distressing material (Reference Fischer, Wik and FredriksonFischer et al, 1996).
A replicated finding has been the deactivation of Broca's area, the area of the brain thought to be responsible for applying semantic representations to personal experience to allow its communication or description (Reference Rauch, van Der Kolk and FisherRauch et al, 1996; Reference Shin, McNally and KosslynShin et al, 1997b ). This would appear to be consistent with subjects with PTSD having difficulty in cognitively restructuring their traumatic experience.
The modality of re-experiencing phenomena will have a bearing on regional brain activation. The mental imagery in the study by Rauch et al (Reference Rauch, van Der Kolk and Fisher1996) was predominantly visual, causing increased rCBF in the secondary visual cortex, a finding not found by Shin et al (Reference Shin, Kosslyn and McNally1997a ,Reference Shin, McNally and Kosslyn b , Reference Shin, McNally and Kosslyn1999) because the re-experiencing phenomena in their study were predominantly tactile. These findings serve to illustrate that different types of trauma cause different emotional reactions; trauma memories are not uniform and activation of brain regions will vary. Further, hypoperfusion of the left inferior frontal gyrus in PTSD (Shin et al Reference Shin, Kosslyn and McNally1997a ,Reference Shin, McNally and Kosslyn b , Reference Shin, McNally and Kosslyn1999) may reflect the nature of intrusive thoughts because the frontal regions are implicated in effortful recall (Reference Schacter, Alpert and SavageSchacter et al, 1996), whereas intrusive phenomena are effortless and may therefore lead to hypoperfusion.
DISCUSSION
Neuroimaging is a powerful method to examine the links among structural and functional brain changes, psychopathology and findings from other neurobiological research in traumatised individuals. However, neuroimaging in PTSD is only just emerging from its infancy, with only 30 published reports as of August 2001. Results are far from uniform but the degree of consensus at this early stage is encouraging. Research in the trauma field is fraught with difficulties; one inherent difficulty in all trauma research is the choice of population to be studied. Research studies have tended to examine homogeneous and highly selected trauma populations. Eighteen of these reports (67%; 18/30) were conducted solely on combat veterans and 73% (254/356) of all scanned subjects were male. Furthermore, combat veterans (58%; 219/377) and survivors of childhood physical and sexual abuse (27%; 101/377) accounted for 85% of all subjects recruited to neuroimaging studies. It is vital that future research into the neurobiology of PTSD attempts to incorporate a wider spectrum of trauma.
The triggers causing intrusive thoughts or flashbacks are personal and often idiosyncratic. Research provocation paradigms need to be able to reflect this and if they do not then researchers may be studying different phenomena. The similarity between generalised combat sounds and a self-generated narrative of a fatal motor vehicle accident in which an individual was at fault have little in common and may explain why there was a failure to show amygdalar activation when generalised combat sounds were used rather than personalised trauma scripts. To expect uniform activation of the brain in different trauma populations with varying provoking stimuli is to suggest that all traumas have similar effects and trigger the same symptoms.
The failure of studies to include control or comparison groups (of trauma-exposed subjects without PTSD) leaves the possibility that the observed changes are due to trauma exposure as opposed to PTSD itself. However, there are demonstrated biological changes following trauma exposure that are associated with PTSD and not simply with the trauma exposure per se (Reference Yehuda and McFarlaneYehuda & McFarlane, 1995). The neurobiology of PTSD is a progressive state of modification and a cross-sectional perspective cannot answer some of the fundamental questions. Could a pre-existing brain abnormality predispose to PTSD or to exposure to trauma, thereby identifying at-risk individuals? Alternatively, could the trauma exposure rather than PTSD be responsible for the demonstrated structural and functional changes (Reference SapolskySapolsky, 2000)? Only one study has examined neuroimaging changes in individuals with acute PTSD and this suggests that no abnormality existed prior to the development of PTSD (Reference Bonne, Brandes and GilboaBonne et al, 2001). Furthermore, are there identifiable trait effects (defining the underlying disease process) as opposed to state effects (reflecting the symptom severity)? Only the latter have been investigated so far in PTSD.
Other methodological issues would include the standardisation of image acquisition (and analysis) and a consensus on methodologies (e.g. type of trauma population, type of symptom provocation and method of diagnosis). In addition, studies controlling for past and current treatment and the presence or absence of comorbidity are needed to determine the relative contributions. This would allow the pooling of data or a meta-analysis that would compensate for individually small studies.
Neuroimaging studies in PTSD have suggested a number of brain regions meriting further attention. Key regional abnormalities, their replicability and the possible significance of each finding are described in Table 6. Findings to support the proposed right-hemisphere lateralisation of post-trauma symptoms are inconsistent, with no neuroimaging study in PTSD examining the role of dominance (studies have chosen to study dextrals) or gender on this proposed laterality. For example, gender differences have been demonstrated in studies of self-induced dysphoria (Reference Whalen, Rauch and EtcoffWhalen et al, 1998b ), with bilateral activation in women and predominantly left-sided activation in men.
Table 6 Central findings of neuroimaging studies

| Finding | Replicability1 |
|---|---|
| Decreased hippocampal volume | +++2 |
| Increased amygdala activity | +++2 |
| Decreased Broca's area (left inferior cortex) activity | +2 |
| Hemispheric lateralisation | +++2 |
| Decreased anterior cingulate cortex activation | ++2 |
| Decreased N-acetyl aspartate in medial temporal regions | + |
| Activation in visual cortex | +2 |
Treatment implications
Neuroimaging findings suggest that after psychological trauma biological changes are not restricted to dysregulation of neurochemical systems but involve alterations in brain function and structure. The challenge for clinicians is to employ therapies for patients with PTSD that prevent, halt or reverse these changes. Functional brain changes after successful treatment have been demonstrated in other conditions (Reference Schwartz, Stoessel and BaxterSchwartz et al, 1996) and preliminary data suggest that the same is true for patients with PTSD (Reference Levin, Lazrove and van der KolkLevin et al, 1999). One possible target is the demonstrated hippocampal damage. The hippocampus may be unique in the brain in its ability to regenerate neurons (Reference Gould, Tanapat and McEwenGould et al, 1998), with agents such as phenytoin potentially able to reverse stress-activated hippocampal atrophy (Reference Watanabe, Gould and CameronWatanabe et al, 1992). Silver et al (Reference Silver, Shin and McNamara1991) have suggested that anticonvulsants may reduce limbic kindling in PTSD, thereby preventing progression of symptoms, but this has yet to be tested in clinical trials in subjects with acute post-traumatic reactions.
Perhaps one of the least expected early findings is the hypoperfusion of Broca's area when trauma-related memories are provoked. Broca's area is necessary for the labelling of emotions, therefore its deactivation under symptom provocation would explain why patients with PTSD can experience intense emotions without being able to label and understand them. This marries clinically with survivors often describing an inability to put their experience into words — it is, in effect, ‘unspeakable’. Therefore, the ability of psychological treatment, especially ‘talking therapies’, may be compromised during some phases of the disorder. Therapies incorporating exposure have proven efficacy for the treatment of PTSD (Reference van Etten and Taylorvan Etten & Taylor, 1998) and this may be because they can target all sensory modalities and not just their semantic representations. Alternatively, their potency may be explained by the preclinical finding that the reactivation of memory allows its disruption (Reference Nader, Schage and Le DouxNader et al, 2000). Importantly, the reactivation of memory does not require it to be put into communicable language
The strategies and findings of published neuroimaging studies in PTSD provide a framework for future research, not just in neuroimaging but for clinical trials of historically accepted treatments for trauma survivors. Future neuroimaging studies need to develop protocols to investigate state and trait effects in a range of traumatic events and to study treatment-naïve subjects. Pre— and post-treatment studies also need to be completed to assess the full effectiveness of clinical strategies.
CLINICAL IMPLICATIONS
-
• More work is needed to identify treatments that reverse the demonstrated functional and structural regional brain changes.
-
• The effectiveness of psychological treatments, especially ‘talking therapies’, may be compromised by underactivity of Broca's area when individuals are accessing traumatic material.
-
• Possible right-hemispheric lateralisation may explain the ‘timeless’ quality of traumatic memories.
LIMITATIONS
-
• The majority of studies are restricted to highly selected and homogeneous samples.
-
• Many of the studies lacked a control or comparison group and therefore did not exclude the possibility that observed changes are the result of exposure to the trauma itself rather than to post-traumatic stress disorder.
-
• Unpublished studies are not discussed.
Acknowledgements
I would like to thank Professors David A. Alexander and Ian C. Reid for their assistance in revising previous drafts of this paper, and Isla Imrie and Louise Winning for locating many of the articles.







eLetters
No eLetters have been published for this article.