


Social phobia or anxiety disorder is increasingly recognised as a highly prevalent and chronic disorder with onset during the teenage years (Reference Lépine and PélissoloLépine & Pélissolo, 1996; Reference Wittchen, Stein and KesslerWittchen et al, 1999). Although the disorder is associated with significant disability (including educational and occupational) which has a negative impact on quality of life, it is both underdiagnosed and undertreated (Reference KasperKasper, 1998). Early work demonstrated that monoamine oxidase inhibitors (e.g. phenelzine) were effective in the treatment of the disorder, but these agents are limited by their side-effect profile, the need for dietary precautions, and drug interactions (Reference VersianiVersiani, 2000). More recent work has established the efficacy of several selective serotonin reuptake inhibitors (SSRIs) (Reference Stein, Fyer and DavidsonStein et al, 1999; Reference Van Ameringen, Lane and WalkerVan Ameringen et al, 2001; Reference Liebowitz, Stein and TancerLiebowitz et al, 2002) and these agents have been recommended as first-line pharmacotherapy (Reference Ballenger, Davidson and LecrubierBallenger et al, 1998). This study investigates the efficacy and tolerability of escitalopram in the treatment of generalised social anxiety disorder.
METHOD
Study design and dosing schedule
This multinational study was a randomised, parallel group, placebo-controlled trial involving 41 centres in eight countries (Austria, Canada, Denmark, Finland, Germany, Norway, South Africa and the UK). Patients who met selection criteria entered a 1-week, single-blind, placebo lead-in period before being randomised to 12 weeks of double-blind treatment with escitalopram or matched placebo capsules. Patients were contacted for a safety follow-up 30 days after their last dose. The initial dosage of escitalopram was 10 mg per day. The dosage could be increased to 20 mg per day after 4, 6 or 8 weeks of treatment in case of an unsatisfactory response, judged as a score above 5 on the Clinical Global Impression scale rating for severity (CGI–S; Reference GuyGuy, 1976) or no decrease in CGI–S score since baseline. The mean daily dose of escitalopram was 17.6 mg at week 12. Efficacy and tolerability were assessed at baseline and after 1, 2, 3, 4, 6, 8 and 12 weeks of treatment.
Patient population
The patient population comprised female and male out-patients with a primary diagnosis of generalised social anxiety disorder established by means of a diagnostic interview following DSM–IV criteria (American Psychiatric Association, 1994), using the Mini-International Neuropsychiatric Interview (MINI; Reference Sheehan, Lecrubier and SheehanSheehan et al, 1998) to assist in the exclusion of disallowed comorbidity. The patients were mainly recruited through advertisements. At the screening visit, patients 18–65 years old were selected if they had a total score of at least 70 on the Liebowitz Social Anxiety Scale (LSAS; Reference LiebowitzLiebowitz, 1987) with exhibited fear or avoidance traits in at least four social situations, and were otherwise healthy based on a physical examination. Patients were excluded if they had another Axis I disorder that was considered the primary diagnosis within the previous 6 months, if the investigator diagnosed a serious risk of suicide or if the Montgomery–Åsberg Depression Rating Scale (MADRS; Reference Montgomery and ÅsbergMontgomery & Åsberg, 1979) total score was higher than 19. Patients were also excluded if they had a DSM–IV diagnosis of alcohol or drug misuse during the past 6 months, or if they had taken a psychoactive drug (including any type of antidepressant, beta-blocker, benzodiazepine, narcotic, analgesic, antipsychotic or herbal remedy) within 2 weeks (5 weeks for fluoxetine and 6 months for depot neuroleptics) before screening, or if the patient had a positive urine drug screen for opiates, methadone, cocaine, amphetamines or benzodiazepines. The only allowed concomitant use of a psychotropic drug during the study was chloral hydrate taken as a hypnotic but not for more than three consecutive nights. Furthermore, patients with a diagnosis of mania or hypomania, body dysmorphic disorder, schizophrenia/other psychotic disorder, eating disorders, mental retardation or any Axis II cluster diagnosis were also excluded. Patients with a known drug (including citalopram) allergy or hypersensitivity or a known lack of therapeutic response to an adequate trial with citalopram were also excluded. Patients participating in a formal psychotherapy programme that went beyond medical counselling were not included.
Efficacy assessments
The primary efficacy measure was the mean change from baseline to the last assessment (carried forward) of the LSAS total score. This scale consists of 24 items, 13 describing performance situations and 11 describing social interaction situations (Reference LiebowitzLiebowitz, 1987). Each of the items is separately rated for ‘fear’ and ‘avoidance’ using a four-point categorical scale. All investigators attended supervised group sessions in order to standardise the interview and rating techniques. Secondary efficacy measures included:
-
(a) mean change from baseline to each visit in the LSAS sub-scale scores for ‘fear/anxiety’ and ‘avoidance’;
-
(b) CGI–S score per visit and change from baseline to visit;
-
(c) Clinical Global Impression – Improvement (CGI–I) score: proportion of responders to treatment, defined as patients achieving a score of 1 (very much improved) or 2 (much improved) on the CGI–I;
-
(d) Sheehan Disability Scale (SDS; Reference SheehanSheehan, 1983) score, for the three domains ‘work’, ‘social’ and ‘family’;
-
(e) change from baseline to each visit in MADRS total score (the MADRS consists of ten items, each rated on a scale from 0 to 6).
Safety and tolerability
Safety assessments were based on vital signs (in a sitting position after 5 min rest), body weight, clinical laboratory tests (including haematology and biochemistry) and electrocardiograms (ECGs), and were assessed at the screening visit and at week 12. Adverse events observed by the investigator, spontaneously reported by the patient or reported in response to nonleading questions were recorded at each visit. The investigator documented the relationship to treatment, onset duration and intensity (mild, moderate or severe). All adverse events were coded using the included term according to the World Health Organization Adverse Reaction Terminology.
Statistical analysis
Efficacy analyses were based on the full analysis set (corresponding to the intent-to-treat population), which comprised all randomised patients who took double-blind study product and had at least one valid post-baseline assessment of the primary efficacy measure. Safety analyses were based on the set of all patients treated, which included all patients who took at least one dose of double-blind study product.
A minimum of 135 patients per treatment arm was required to reach a power of 90% to detect a significant difference between treatment groups in mean change from baseline to final assessment in LSAS total score at the 5% significance level. A general linear model for analysis of covariance (ANCOVA) was applied to the primary and secondary efficacy measures with factors for treatment group and centres (all centres with fewer than four patients were collapsed into one collective centre), and with baseline LSAS total score as a covariate. The final CGI–S and CGI–I scores were also analysed using the non-parametric Cochran–Mantel–Haenszel mean score statistics. Between-group comparisons of the proportion of responders (CGI–I score of 1 or 2) to treatment were performed using chi-squared and Fisher's exact tests. Descriptive statistics were used for absolute values and mean changes from baseline in laboratory values, ECG parameters, vital signs and body weight. All statistical tests were two-sided and were carried out at the 5% level of significance.
RESULTS
Patient baseline characteristics
A total of 358 patients were randomised into the study, 177 to placebo treatment and 181 to escitalopram treatment. Of these, 5 patients did not receive double-blind treatment. The full analysis set thus consisted of 177 patients in the escitalopram group and 176 patients in the placebo group. A total of 290 patients (81%) completed the study, 145 in each treatment group (Fig. 1). There were slightly more men than women in both treatment groups. Baseline characteristics were similar for the two treatment groups with the exception of age and duration of the disorder, both of which were slightly higher in the escitalopram group (Table 1). No between-group difference was seen for the severity of the disorder, as measured by the baseline LSAS total score and the CGI score. There was no difference with respect to medical history or physiological variables. Comorbidity with depressive symptoms was low, as judged by the baseline MADRS total score and the low number of patients with a diagnosis of comorbid depression or dysthymia (Table 1). The high baseline LSAS total score and the baseline SDS score between 6 and 7 (on a ten-point scale) for the work and social life items are in line with the average CGI–S score, indicating a markedly ill patient population.
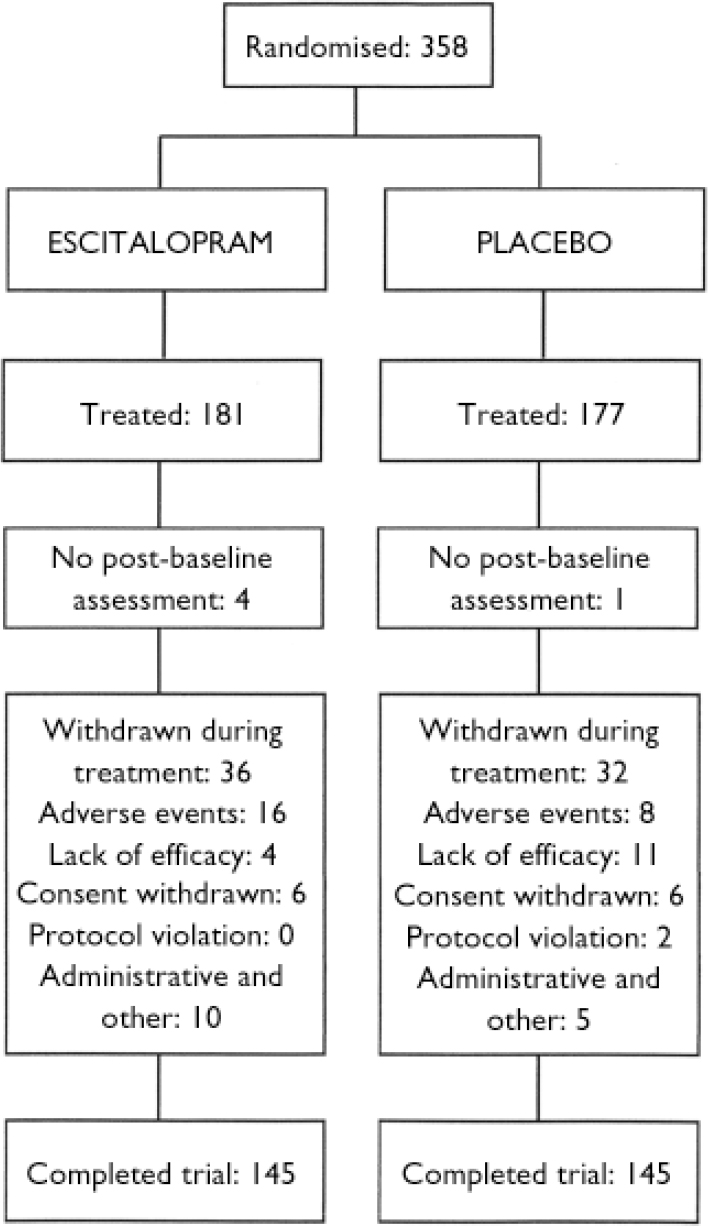
Fig. 1 Study profile.
Table 1 Patient characteristics at baseline assessment
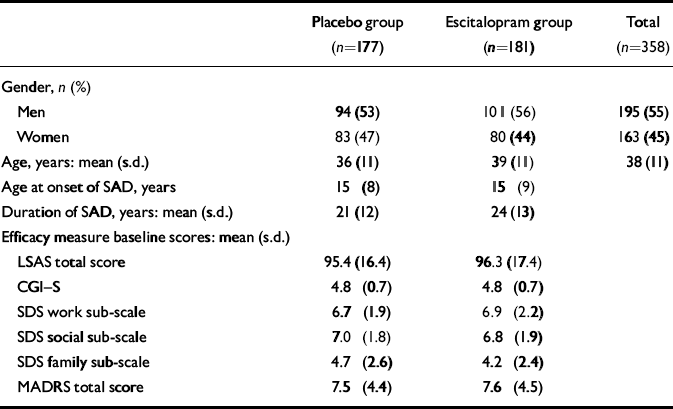
| Placebo group (n=177) | Escitalopram group (n=181) | Total (n=358) | |
|---|---|---|---|
| Gender, n (%) | |||
| Men | 94 (53) | 101 (56) | 195 (55) |
| Women | 83 (47) | 80 (44) | 163 (45) |
| Age, years: mean (s.d.) | 36 (11) | 39 (11) | 38 (11) |
| Age at onset of SAD, years | 15 (8) | 15 (9) | |
| Duration of SAD, years: mean (s.d.) | 21 (12) | 24 (13) | |
| Efficacy measure baseline scores: mean (s.d.) | |||
| LSAS total score | 95.4 (16.4) | 96.3 (17.4) | |
| CGI–S | 4.8 (0.7) | 4.8 (0.7) | |
| SDS work sub-scale | 6.7 (1.9) | 6.9 (2.2) | |
| SDS social sub-scale | 7.0 (1.8) | 6.8 (1.9) | |
| SDS family sub-scale | 4.7 (2.6) | 4.2 (2.4) | |
| MADRS total score | 7.5 (4.4) | 7.6 (4.5) |
Patient withdrawals
A total of 68 patients (19%) withdrew from the study, with no overall between-group difference (18% in the placebo group and 20% in the escitalopram group). However, numerically more patients in the escitalopram group (8.8%) than in the placebo group (4.5%) withdrew because of adverse events and numerically more patients in the placebo group (6.2%) than in the escitalopram group (2.2%) withdrew because of lack of efficacy, with the latter difference approaching statistical significance (P=0.059).
Efficacy results
Primary efficacy outcome
During double-blind treatment the LSAS total score decreased in the escitalopram group from a baseline value of 96.3 (s.d.=17.4) to 62.2 (s.d.=30.7) at week 12 (last observation carried forward; LOCF) and in the placebo group from 95.4 (s.d.=16.4) to 68.8 (s.d.=29.7). The treatment difference of 7.3 between escitalopram and placebo in change from baseline to week 12 in favour of escitalopram was statistically significant (ANCOVA, P=0.005) (Fig. 2). Exploratory analyses of potential covariates revealed no treatment-by-centre or treatment-by-baseline LSAS total score interaction effect. The same was true for treatment interactions with gender, age and duration of disorder.
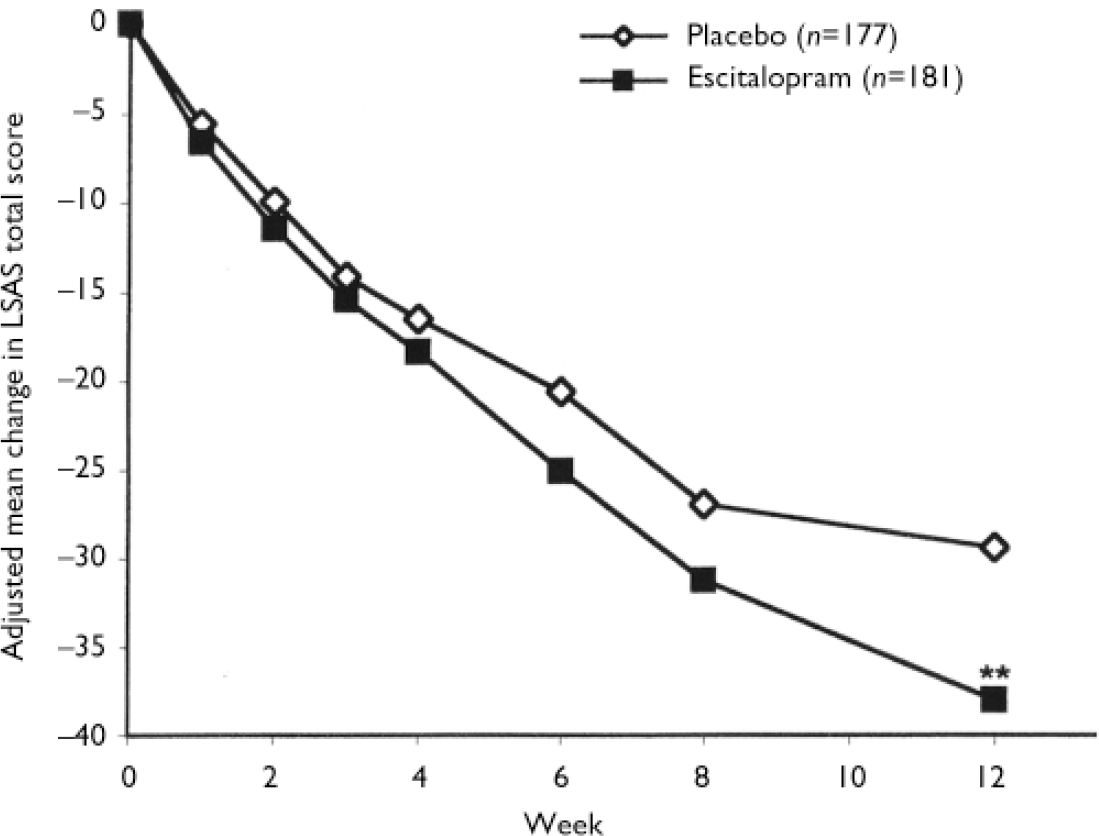
Fig. 2 Mean change from baseline in Liebowitz Social Anxiety Scale (LSAS) total score (last observation carried forward; LOCF) by week, for the escitalopram and placebo groups (full analysis set), adjusted for baseline score and centre by least squares mean analysis of covariance (** P<0.01v. placebo).
Secondary efficacy measures
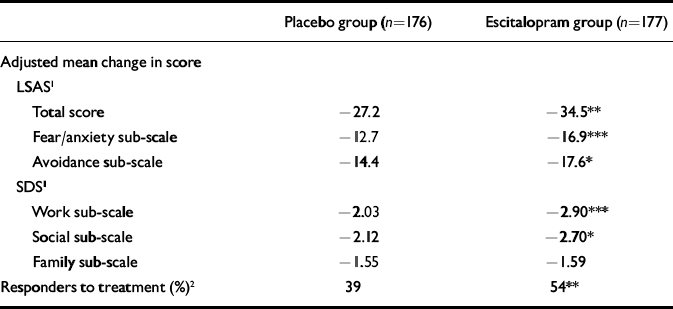
The mean change from baseline to end-point (LOCF) in the LSAS sub-scale scores was statistically significant in favour of escitalopram at week 12 (P<0.05) for ‘avoidance’ and at weeks 6 and 12 (P<0.001) for ‘fear/anxiety’, but not for the SDS ‘family’ sub-scale (Table 2). Superiority of escitalopram over placebo was also manifested in the change from baseline to week 12 (LOCF) in CGI–S score (P<0.01), the mean CGI–I score at week 12 (P<0.001) and in the change from baseline to week 12 (LOCF) in the two SDS items ‘work’ (P=0.01) and ‘social’ (P=0.02) (Table 2).
Table 2 Adjusted mean change from baseline to week 12 in LSAS and SDS scores and response rate
| Placebo group (n=176) | Escitalopram group (n=177) | |
|---|---|---|
| Adjusted mean change in score | ||
| LSAS1 | ||
| Total score | -27.2 | -34.5** |
| Fear/anxiety sub-scale | -12.7 | -16.9*** |
| Avoidance sub-scale | -14.4 | -17.6* |
| SDS1 | ||
| Work sub-scale | -2.03 | -2.90*** |
| Social sub-scale | -2.12 | -2.70* |
| Family sub-scale | -1.55 | -1.59 |
| Responders to treatment (%)2 | 39 | 54** |
A total of 54% of escitalopram-treated patients and 39% of placebo-treated patients responded to treatment (LOCF, P<0.01). The corresponding figures for the observed case (OC) analysis were 63% of escitalopram-treated patients and 43% of placebo-treated patients (P<0.001).
Safety results
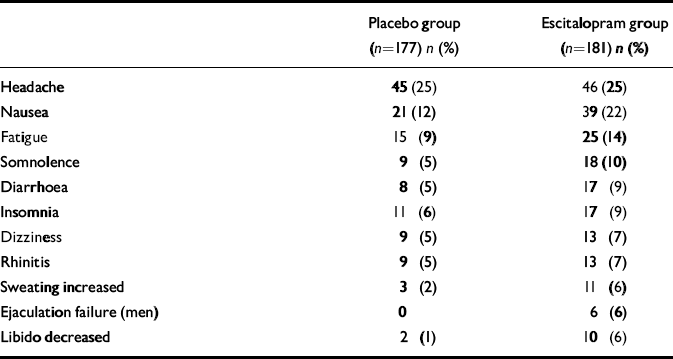
Table 3 shows all treatment-emergent adverse events with an incidence of more than 5% in either treatment group. No clinically relevant trend was observed in mean ECG or in clinical laboratory parameters.
Table 3 Treatment-emergent adverse events with an incidence greater than 5%
| Placebo group (n=177) n (%) | Escitalopram group (n=181) n (%) | |
|---|---|---|
| Headache | 45 (25) | 46 (25) |
| Nausea | 21 (12) | 39 (22) |
| Fatigue | 15 (9) | 25 (14) |
| Somnolence | 9 (5) | 18 (10) |
| Diarrhoea | 8 (5) | 17 (9) |
| Insomnia | 11 (6) | 17 (9) |
| Dizziness | 9 (5) | 13 (7) |
| Rhinitis | 9 (5) | 13 (7) |
| Sweating increased | 3 (2) | 11 (6) |
| Ejaculation failure (men) | 0 | 6 (6) |
| Libido decreased | 2 (1) | 10 (6) |
DISCUSSION
Patient population
The typical onset of social anxiety disorder during adolescence, with its chronic course, its high level of psychiatric comorbidity and its low spontaneous remission rate, contributes to serious impairment of daily functioning in the professional and social life of those with this disorder (Reference Lépine and PélissoloLépine & Pélissolo, 2000). These epidemiological characteristics were reflected among our participants. The mean age of onset was 15 years and the chronicity of the disorder was evident from its average duration, which was more than 20 years. Sheehan Disability Scale mean baseline scores for ‘work’ and ‘social’ items (around 7 on the ten-point sub-scales) indicate the negative impact of the disorder on daily life functioning in this group.
In order to investigate the specific efficacy of escitalopram in the treatment of social anxiety disorder, the study selected a somewhat atypical patient population with a low level of comorbidity. The average MADRS total score of 7.5 indicates the absence of significant depressive symptoms. It can thus be concluded that the patient population in this study represents patients with relatively pure, generalised social anxiety disorder. The average LSAS total score at baseline of over 95 indicates a more severely ill patient population than that in other published clinical drug trials (Reference Baldwin, Bobes and SteinBaldwin et al, 1999; Reference Liebowitz, Stein and TancerLiebowitz et al, 2002).
Therapeutic efficacy and placebo response
This study of the SSRI escitalopram confirmed the efficacy of SSRIs in the treatment of generalised social anxiety disorder. Escitalopram had a significantly better effect than placebo at the end of the 12-week trial period on both the primary and the secondary efficacy measures, including the two LSAS sub-scales of ‘fear/anxiety’ and ‘avoidance’. The primary analysis showed a decrease in the total LSAS score of 34.4 points in the escitalopram group and a relatively large decrease of 27.2 points in the placebo group. The effect size in the escitalopram group is comparable with that reported in other studies of SSRIs in the treatment of generalised social anxiety (Reference Stein, Liebowitz and LydiardStein et al, 1998; Reference AlluganderAllugander, 1999; Reference Baldwin, Bobes and SteinBaldwin et al, 1999). However, no other published study has reported a placebo response as high as 39% (LOCF) in social anxiety disorder. A review by Oosterbaan et al (Reference Oosterbaan, van Balkom and Spinhoven2001) analysed 15 placebo-controlled studies and concluded that a moderate placebo response is seen in this disorder which appears to be lower than that in depression or panic disorder. The review found no evidence of an increase in the placebo response in studies of social anxiety over the past decade, although this is seen for other disorders. There was, however, a trend towards a higher response rate in the placebo groups, but not in the active treatment groups, with increasing sample size. No relation was found between the baseline severity of social anxiety disorder and improvement during treatment, as measured by the mean change from baseline or the percentage of responders. This is somewhat in contrast to other studies of this disorder, in which placebo responders were generally less symptomatic (Reference MontgomeryMontgomery, 1998) and where a better separation between active medication and placebo was seen among the more severely affected patients. The trend towards a higher response rate in the placebo groups with increasing size of trial, as found by Oosterbaan et al (Reference Oosterbaan, van Balkom and Spinhoven2001), is consistent with the substantial size of our trial.
Irrespective of the placebo response size, the clinical significance of the escitalopram treatment effects in this study was demonstrated by statistically significant effects on the global measures of severity of illness and improvement (CGI–S and CGI–I) and, importantly, also on the two Sheehan Disability Scale items ‘work’ and ‘social’. A final score on the CGI–I scale of 1 or 2 (very much or much improved) has commonly been used as a response criterion in social anxiety disorder pharmacotherapy trials. In this trial, the escitalopram response rate (OC) was 63% compared with 43% in the placebo group. Again, the magnitude of response of the escitalopram-treated patients is consistent with that reported in other studies, whereas the placebo response rate is higher than that found previously (Reference Liebowitz, Stein and TancerLiebowitz et al, 2002).
Withdrawals
The total withdrawal rate of 19% is clearly lower than that in a recently reported fixed-dose study with paroxetine (Reference Liebowitz, Stein and TancerLiebowitz et al, 2002) and somewhat lower than the average rate of 23% based on the 15 studies reviewed by Oosterbaan et al (Reference Oosterbaan, van Balkom and Spinhoven2001). The latter review further reported a positive relation between withdrawal rate and the size of the trials. The withdrawal rates varied slightly between treatment groups in our study, with borderline statistical significance for the higher rate of withdrawals due to lack of efficacy in the placebo group, and a somewhat higher withdrawal rate due to adverse events in the escitalopram group.
Tolerability
Escitalopram was well tolerated in this study, with prevalence rates of single adverse symptoms comparable with those in studies of its use in depression (Reference Wade, Lemming and HedegaardWade et al, 2002). A favourable tolerability profile is important in the pharmacotherapy of this chronic disease, for which lengthy treatment may be required. Headache was the adverse event with the highest incidence, and its incidence was similar in the two treatment groups. Nausea, increased sweating and sexual side-effects occurred with a higher incidence during escitalopram treatment. Also in agreement with depression studies, no clinically relevant mean change in ECG variables was seen in the escitalopram group.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
▪ Escitalopram is efficacious in the treatment of generalised social anxiety disorder.
-
▪ The tolerability of escitalopram in the dosage range 10–20 mg was as favourable as that previously seen in the treatment of depression.
-
▪ The efficacy/tolerability profile of escitalopram and the low withdrawal rate in this study make escitalopram a valuable pharmacotherapeutic option in the treatment of patients with social anxiety disorder.
LIMITATIONS
-
▪ A high placebo response rate was found in this study.
-
▪ Given the chronic course of the disease, studies with a longer treatment duration may be warranted to assess further potential improvements.
-
▪ The patient sample was selected to minimise comorbidity with other psychiatric disorders in order to investigate effects of escitalopram specifically in social anxiety disorders.
Acknowledgements
The authors acknowledge the valuable contributions of all participating. investigators in this study. Austria: Siegfried Kasper, Hans Georg. Zapotoczky, Anton Tölk, Albert Wuschitz; Canada: Michael Van. Ameringen, Jacques Plamondon, Kevin Dwight Kjernisted, Pratap Rao Chokka, Stanley P. Kutcher, Martin A. Katzman; Denmark: Henrik Lublin, Eva. Jensen, Annemarie Lund Laursen, Peter Kaare Østergaard, Flemming. Bjørndal, Palle Stensbæk Schrøder; Finland: Hannu. Juhani Koponen, Anna Eliisa Savela, Anneli Timonen, Ulla Marjatta Lepola, Riitta Helena Jokinen, Markus Pulkkinen; Germany: Dolores Backhaus, Johannes Böhringer, Karl Ditzler, P. Donat, Buckhard Flötotto, Hinrich Hörnlein-Rummel, Gerhard Roth, Christoph Schenk; Norway: Alv A. Dahl, Fred Holsten; South Africa: Dan J. Stein, William J. C. Verbeeck, Catherine Mary Maud, Dora Wynchank, Donald A. B. Wilson, Premakanthie R. Laban, Wally Landsberg, Chané Magnus; UK: Alan. Wade.
R.N. and H.L. are full-time employees of H. Lundbeck AS. S.K. has received. grants or research support from Eli Lilly, Lundbeck, Bristol-Myers Squibb, GlaxoSmithKline, Organon and Servier. He works or has worked as a consultant. or on an advisory board for AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly, Lundbeck, Pfizer, Organon, Janssen Pharmaceutica. and Novartis. His speaker's bureaux include AstraZeneca, Eli Lilly, Lundbeck and Janssen Pharmaceutica. D.S. has received research grants and/or. consultancy honoraria from AstraZeneca, Eli Lilly, GlaxoSmithKline, Lundbeck, Orion, Pfizer, Pharmacia, Roche, Servier, Solvay, Sumitomo and Wyeth.






eLetters
No eLetters have been published for this article.