


For the past hundred years most clinical work and research in psychiatry has proceeded under the assumption that schizophrenia and bipolar affective disorder (or the corresponding earlier terms, such as dementia praecox and manic–depressive illness) are distinct entities with separate underlying disease processes and treatments. This so-called ‘Kraepelinian dichotomy’ has pervaded Western psychiatry since Emil Kraepelin (Reference Kraepelin and Barclay1919) ‘crystallised dementia praecox and manic–depressive illness from an amorphous mass of madness’ (Reference Brockington and LeffBrockington & Leff, 1979), and remains enshrined in current classifications. However, many individuals with severe psychiatric illness have both prominent mood and psychotic symptoms – raising the possibility, indeed the likelihood, that there is not a neat biological distinction between schizophrenia and bipolar affective disorder. Genetic epidemiology has always been influential in shaping and validating psychiatric nosology (Reference Robins and GuzeRobins & Guze, 1970). Now molecular genetic studies are beginning to challenge and will soon, we predict, overturn the traditional dichotomous view.
WHY HAS THE KRAEPELINIAN DICHOTOMY SURVIVED FOR SO LONG?
In the absence of ‘laboratory’ tests based on a solid understanding of pathogenesis, the criteria available to psychiatry for validating nosological categories have been restricted to clinical features, outcome and family history (Reference Robins and GuzeRobins & Guze, 1970). These were the tools used by Kraepelin in formulating his ideas and have been applied to research data in shaping the modern operational classifications. One of the key scientific observations supporting the Kraepelinian dichotomy was that the prototypical disorders tend to ‘breed true’. Thus, a consistent finding has been a substantially increased risk of schizophrenia but not bipolar disorder in the relatives of probands with schizophrenia, and vice versa in corresponding studies of bipolar disorder. It is also true that groups of individuals classified as having typical schizophrenia can be discriminated from sets of individuals classified as having typical bipolar disorder on the basis of clinical features and outcome.
As well as having apparent empirical support, the Kraepelinian view holds attractions for clinicians; it is conceptually simple and allows psychiatrists to demonstrate diagnostic expertise by exercising judgement over an often complex clinical picture and to reach a clear diagnosis. However, most psychiatrists, although willing to make use of the advantages of the dichotomy, are fully aware of its limitations, and this is mirrored in the failure of nosologists to identify any ‘point of rarity’ between the two disorders (Reference KendellKendell, 1987). Cogent arguments for abandoning a categorical approach in favour of a dimensional or continuous formulation have been advanced (e.g. Reference CrowCrow, 1990). However, these failed to gain widespread support, in part because of a lack of robust scientific data, and possibly also because of the practical complexity of applying dimensional classifications in clinical practice and research settings.
WHY IS THIS DICHOTOMY NOW BEING CHALLENGED?
Evidence from genetic epidemiology has been gradually accumulating over the past two decades that is inconsistent with the dichotomous view, and recent molecular genetic findings seem set finally to overturn it. Key pieces of evidence include the following.
-
(a) Family studies point to the existence of a non-trivial degree of familial coaggregation between schizophrenia and bipolar illness and between schizo-affective disorders and both bipolar disorder and schizophrenia (reviewed by Reference Craddock, O'Donovan and OwenCraddock et al, 2005).
-
(b) A recent twin study – the only one that has used an analysis unconstrained by the diagnostic hierarchy inherent in current classification systems – demonstrated an overlap in the genetic susceptibility to mania and schizophrenia (Reference Cardno, Rijsdijk and ShamCardno et al, 2002) and provided evidence that there are genes that confer susceptibility across the Kraepelinian divide, to schizoaffective disorder and to some cases of schizophrenia and bipolar disorder. This study also confirmed the traditional notion that there are genes specific to the two prototypical disorders.
-
(c) Systematic, whole-genome linkage studies of schizophrenia and bipolar disorder have implicated some chromosomal regions in common; this is consistent with the presence of shared susceptibility genes (Reference BerrettiniBerrettini, 2003; Reference Craddock, O'Donovan and OwenCraddock et al, 2005).
-
(d) Most recently, and most convincingly, genes have been identified in which variation appears to confer risk to both schizophrenia and bipolar disorder. One example is the gene encoding d-amino acid oxidase activator (formerly known as the G72/G30 locus) on chromosome 13q, one of the regions implicated in genome scans of both disorders (Reference Craddock, O'Donovan and OwenCraddock et al, 2005). This locus was originally reported as showing association in schizophrenia in two independent samples. Subsequently association has been reported in bipolar disorder in three independent samples. Another example is the gene Disrupted in Schizophrenia 1 (DISC1). The gene, as the name implies, is disrupted in a family in which both schizophrenia and bipolar disorder co-segregate with a chromosomal translocation. Recent findings suggest that schizophrenia, schizoaffective disorder and bipolar disorder might be associated with polymorphisms in this gene (Reference Craddock, O'Donovan and OwenCraddock et al, 2005).
WHAT ARE THE IMPLICATIONS FOR PSYCHIATRIC RESEARCH?
The Kraepelinian dichotomy has served academic psychiatry well. Indeed, Kraepelinian diagnoses formed the basis of recent successes in genetics, probably because their net effect is to simplify the genetic architecture of the groups defined, albeit at the expense of excluding many cases. The dichotomy also formed the basis of the operational diagnostic criteria that brought a degree of rigour and reproducibility to psychiatric research. However, there is a danger that it will now impede rather than aid progress. The recent findings are compatible with a model of functional psychosis in which susceptibility to a spectrum of clinical phenotypes is under the influence of overlapping sets of genes which, together with environmental factors, determine an individual's expression of illness (Fig. 1). Such a model, although a better approximation than the dichotomous view, is itself only crude. A more accurate model would probably be based in multidimensional space because, in addition to the interface between bipolar disorder and schizophrenia, there is genetic overlap between the functional psychoses and major depressive disorder – and, indeed, other disorders – with extension into subclinical (or normal) variation. It seems likely that sets of overlapping genes will be identified that confer risks along different domains of psychopathology, corresponding to the disruption of different brain systems. Unravelling the biology underlying these overlaps will shed light on the bewildering degree of ‘comorbidity’ observed across disorders and the widespread non-specificity of treatments.
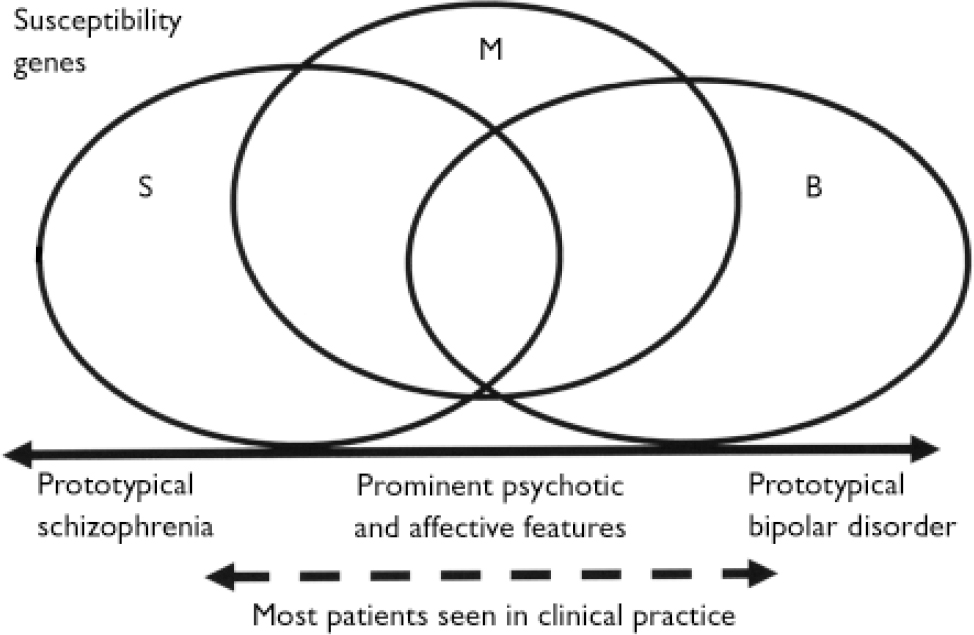
Fig. 1 Possible relationship between susceptibility genes and the clinical picture for disorders in the psychosis–bipolar spectrum. Recent genetic studies suggest that there are genes specific to schizophrenia (S), genes specific to bipolar disorder (B) and genes that confer risk to schizoaffective disorder, schizophrenia and bipolar disorder (M). The combination of susceptibility genes inherited by an individual, together with the environmental exposures, determine the key clinical features of the illness, positioned on a spectrum from prototypical schizophrenia at one end to prototypical bipolar disorder at the other. Most cases lie somewhere in the central part of the spectrum.
This research agenda will best be served by adopting broader inclusion criteria for the functional psychoses and by a combination of inductive and hypothesis-driven approaches aimed at relating biological processes to symptoms and syndromes defined at both clinical and endophenotypic levels. This will require more detailed clinical analysis and the integration of data across multiple domains such as genetics, environmental measures, brain imaging and cognitive neurosciences. In order to achieve this, academic psychiatry will need to scale up its ambitions and plan detailed multidisciplinary, multicentre studies of large numbers of individuals with psychosis. The creation of the Mental Health Research Network under the auspices of the UK Clinical Research Collaboration offers a possible route towards such studies in the UK.
Genetics will have an increasingly important role in all research aimed at understanding the aetiology and pathogenesis of psychosis. As risk genes are identified, so it will become possible to determine how genetic variation relates to clinical variation across and outwith current diagnostic categories, and to explore the relationship between variation in specific susceptibility genes and the disruption of functional systems using techniques such as imaging, psychological testing and neuropathological studies. Thus it will become increasingly possible to seek correlations between psychopathology and biological dysfunction. For example, genetic risk for prototypical schizophrenia might in part be mediated by neurodevelopmental abnormalities with associated structural brain changes and cognitive impairments (Reference Murray, Sham and van OsMurray et al, 2004). Risk of developing positive psychotic symptoms might be conferred by genetically influenced abnormalities in dopamine and glutamate neurotransmission, with abnormalities of synaptic function leading to abnormal connectivity (Reference Owen, O'Donovan and HarrisonOwen et al, 2005). These suggestions are illustrative, to indicate the directions that research is likely to take in the coming decade and the power of genetics to shape this agenda. Epidemiology, too, will benefit from integration of the analysis of genetic and environmental risk factors and exploration of the interplay between these two classes of aetiological agent.
WHAT ARE THE IMPLICATIONS FOR CLINICAL PRACTICE?
In the coming years psychiatrists are likely to have at their disposal simple and inexpensive tests to help identify the pathways involved in an individual's illness and there-by inform treatment decisions. Such tests will not replace the clinical skills now used in diagnosis and management but will be tools to aid these processes, much as lipid levels and blood enzyme measurements aid cardiologists in management of cardio-vascular disease.
Changes in classification will accompany the improvements in understanding of pathogenesis. These will require clinicians to embrace classifications that are both more complex (more categories or, perhaps, dimensions) and also simpler (because they map on to the biology of the illness more closely). These developments have much to offer patients and the professional standing of psychiatry. Most patients want to be given an unambiguous and accurate diagnosis, but psychiatrists are understandably reluctant to be too dogmatic in the early stages of psychotic illness, recognising that the cross-sectional picture may change longitudinally – often frustrating patients, leading to diagnostic revisions between categories and creating an impression that psychiatrists are indecisive or incompetent. Moving to a spectrum concept (be it with categories or dimensions) with recognition of overlapping pathogenetic factors and varying expression (dependent upon both genetic risk and environmental exposure) would allow a confident and clear diagnosis to be offered (perhaps ‘psychosis-spectrum illness’ or ‘mood–reality disorder’), with a clear explanation that some specific tests and a period of observation will help to clarify the likely course of illness and response to treatment. This would be greatly preferable to the current situation and the inevitable consequences of damage to the therapeutic alliance caused by diagnostic revisions.
The Kraepelinian dichotomy has been useful for a hundred years. Now it is time to move on.


eLetters
No eLetters have been published for this article.