


Bipolar disorder is a debilitating illness characterised by drastic swings in mood, energy and functional ability. Effective and rapid control of acute mania is required to prevent potential harm to patients and their families. Mood stabilisers and conventional antipsychotics, either alone or in combination, have been the mainstay of treatment in the manic phase of bipolar disorder. The atypical antipsychotics risperidone, olanzapine and quetiapine, when administered as monotherapy (Reference Segal, Berk and BrookSegal et al, 1998; Reference Tohen, Sanger and McElroyTohen et al, 1999; Reference Vieta, Brugue and GoikoleaVieta et al, 2004) or in combination with mood stabilisers (Reference Ghaemi, Sachs and BaldassanoGhaemi et al, 1997; Reference Suppes, Webb and PaulSuppes et al, 1999; Reference Miller, Yatham and LamMiller et al, 2001; Reference Vieta, Goikolea and CorbellaVieta et al, 2001; Sachs et al, Reference Sachs, Grossman and Ghaemi2002, Reference Sachs, Chengappa and Suppes2004; Reference Tohen, Chengappa and SuppesTohen et al, 2002; Reference Yatham, Grossman and AugustynsYatham et al, 2003), have been shown to be effective and well tolerated in the treatment of mixed and manic episodes of bipolar disorder. This study evaluated risperidone monotherapy in the treatment of hospitalised patients with severe acute mania.
METHOD
Trial design
A 3-week, randomised, double-blind trial was conducted at eight sites in India. Patients with a DSM–IV diagnosis of bipolar I disorder (American Psychiatric Association, 1994) who were experiencing a manic or mixed episode and who met the entry criteria were admitted to hospital and then randomly assigned in equal numbers to receive placebo or 1–6 mg of risperidone, initially in a dose of 3 mg once daily, after a wash-out and screening period lasting up to 3 days. Randomisation was stratified by the presence or absence of psychotic features at baseline, manic or mixed episode, and by treatment centre. After randomisation and the initiation of treatment (baseline), patients remained in hospital for at least 7 days. Treatment efficacy was determined primarily by the change from baseline to end-point in mean total scores on the Young Mania Rating Scale (YMRS; Reference Young, Biggs and ZieglerYoung et al, 1978). Most efficacy and safety assessments were made weekly; treatment-emergent adverse events and vital signs were assessed more frequently. One of the sites was withdrawn from the study after enrolling three participants, because of concerns about data quality. The data from these individuals are therefore included only in the safety analyses below.
Participants
In-patients aged 18 years or over were enrolled if they met DSM–IV criteria for bipolar I disorder with either manic or mixed episodes and with or without psychotic features and had a score of at least 20 on the YMRS at screening and at baseline. Signed informed consent was obtained for all participants and the study was conducted according to the Recommendations Guiding Physicians in Biomedical Research Involving Human Subjects, in the 1989 version of the Declaration of Helsinki (World Medical Association, 1989).
Patients with schizoaffective disorder, rapid cycling, borderline or antisocial personality disorder, a recent history of substance dependence, risk of suicide or violent behaviour, and unstable medical illness were excluded. Patients with antidepressant-induced mania or those who had been treated with an antidepressant within 4 weeks of screening were also excluded. Additionally, patients whose YMRS score reduced by 25% or more between screening and baseline were not permitted to enter the study.
Interventions
Patients assigned to receive risperidone were given a single 3 mg dose on day 1. On day 2, at the discretion of the investigator, each patient's once-daily dose (administered in the evening) could be reduced to 2 mg or increased to 4 mg. On day 3, the daily dose could be reduced to 1 mg or increased to 5 mg. On day 4 and thereafter the daily dose could be increased to 6 mg. Throughout the trial, increases in risperidone dosage were made in increments of 1 mg daily and the daily dose could be between 1 mg and 6 mg.
The patients received no psychotropic drug other than risperidone, with the exception of lorazepam. Lorazepam could be given for the control of agitation, irritability, restlessness, insomnia or hostility during the wash-out period and the first 10 days of the double-blind treatment period (≤8 mg per day on days 1 to 3, ≤6 mg per day on days 4 to 7, and ≤4 mg per day on days 8 to 10); however, it was not to be given within 8 h of behavioural assessments. Beta-adrenergic blockers for treatment-emergent akathisia and antiparkinsonian drugs could be given as needed.
Assessments
The primary efficacy variable was the change in total YMRS score from baseline to end-point (last post-baseline observation carried forward to the end of treatment at week 3). The YMRS (scores range from 0 to 60) consists of 11 items; a higher score represents a worse condition. Other measures included the Clinical Global Impression (CGI; Reference GuyGuy, 1976) severity scale (scores range from 0, not ill, to 6, extremely severe); the Global Assessment Scale (GAS; Reference Endicott, Spitzer and FleissEndicott et al, 1976), a measure of functioning (scores range from 0 to 100); the Montgomery–Åsberg Depression Rating Scale (MADRS; Reference Montgomery and AsbergMontgomery & Åsberg, 1979) to measure symptoms of depression (scores range from 0 to 56); and the Positive and Negative Syndrome Scale (PANSS; Reference Kay, Fiszbein and OplerKay et al, 1987) for symptoms of psychosis (PANSS total scores range from 30 to 210). Total scores for PANSS and scores on the five factors (positive symptoms, negative symptoms, disorganised thoughts, uncontrolled excitement/hostility, and anxiety/depression) are reported (Reference Marder, Davis and ChouinardMarder et al, 1997). Severity of movement disorders was assessed by means of the Extrapyramidal Symptom Rating Scale (ESRS; Reference Chouinard, Ross-Chouinard and AnnableChouinard et al, 1980) total score (scores range from 0 to 102). On all but the GAS scale, higher scores indicate more severe symptoms.
Investigators were trained in the use of each of these instruments and certification was required for those administering the YMRS.
Data analysis
The sample size was based on experience from three recent trials of atypical antipsychotic agents in the treatment of bipolar disorder (Reference Tohen, Sanger and McElroyTohen et al, 1999; Reference Sachs, Grossman and GhaemiSachs et al, 2002; Reference Yatham, Grossman and AugustynsYatham et al, 2003). Power calculations were based on a two-sided test with a significance level of 5%. With an anticipated total of 115 patients per group and with a standard deviation of 12.6, the trial would have approximately 90% power to detect a clinically meaningful difference of 5.4 units in the total YMRS score. Since the estimated rate of discontinuation was approximately 20% among the randomly assigned patients, the total number of such patients was set at 288, with 144 per treatment group.
The Van Elteren test and the Cochran–Mantel–Haenszel test were used to analyse continuous and categorical variables, respectively, controlling for centre and psychotic features at baseline. An analysis of covariance (ANCOVA) model was used to analyse the change from baseline in YMRS scores, with the treatment centre and baseline psychosis as factors and the baseline YMRS score as covariate. The main effect for psychosis was included in the model because it was a randomisation stratification factor. Because most of the patients in this trial had purely manic symptoms, the manic or mixed factor was not included in the model, although it was used as a stratification factor. The same method was used to evaluate the changes from baseline in GAS, CGI and MADRS scores.
The primary comparison between the risperidone and placebo treatment groups was the change in YMRS score from baseline based on least-squares means obtained from the ANCOVA model. To determine whether the treatment effects differed according to baseline YMRS score (<30, ≥30), diagnosis (absence v. presence at baseline), age (<23 years, 23–44 years, ≥45 years) or gender, ANCOVA models were fitted separately for each level of these variables. Clinical response, defined as a 50% or greater reduction in YMRS score from baseline, was analysed using a Cochran–Mantel–Haenszel test for general association controlling for centre and psychotic features at baseline. Both observedcase and end-point (last observation carried forward) data are reported.
Adverse events were tabulated and the electrocardiographic data and vital sign scores were compared with prospectively defined abnormality criteria.
RESULTS
Of the 324 patients who were screened, 145 were randomly assigned to placebo and 146 to risperidone. One patient in the placebo group received no treatment (Fig. 1). The baseline characteristics of the two treatment groups were similar (Table 1). No statistically significant or clinically meaningful between-treatment difference on the five measures of psychopathology listed in Table 1 was observed at baseline. A pure manic episode at baseline was observed in 94% of the placebo group and 97% of the risperidone group. The other 12 patients had a mixed episode. Psychotic features were present in 58% of the placebo group and 60% of the risperidone group at baseline. Total PANSS scores were higher in patients diagnosed as having psychotic features, indicating an accurate assessment of the presence of psychosis by the investigators. In the risperidone group, mean PANSS total scores were 58.6 (s.e.=1.0) in patients with psychotic features and 47.4 (s.e.=1.0) in those without psychotic features. The respective scores in the placebo group were 59.0 (s.e.=1.1) and 47.8 (s.e.=1.1).
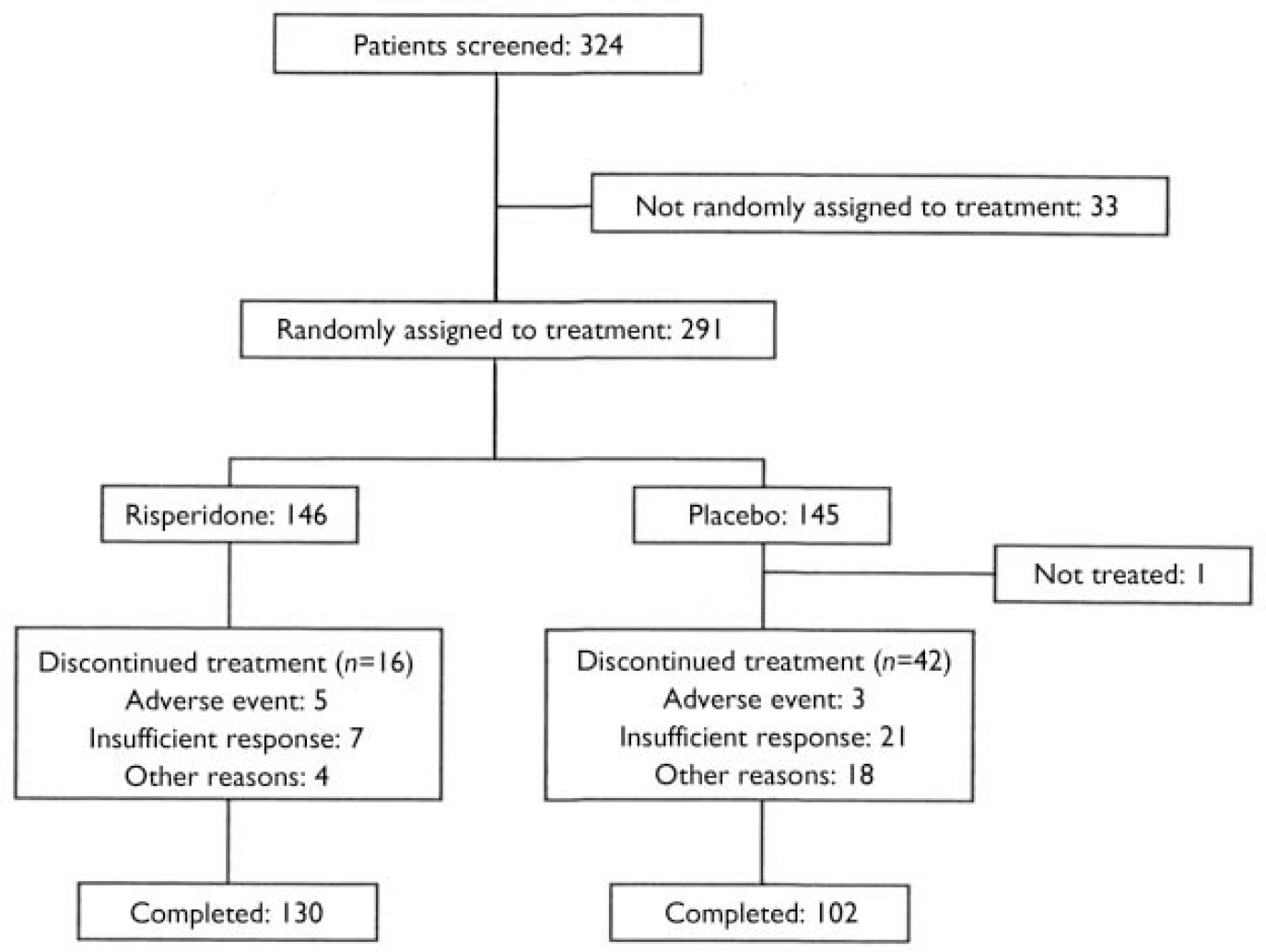
Fig. 1 Study profile.
Table 1 Baseline characteristics of the sample

| Placebo group (n=144) | Risperidone group (n=146) | |
|---|---|---|
| Age, years: mean (s.e.) | 35.5 (1.0) | 34.7 (1.0) |
| Gender, % male | 56 | 68 |
| Body weight, kg: mean (s.e.) | 54.5 (1.0) | 54.4 (0.9) |
| Episode, % | ||
| Manic | 94 | 97 |
| Mixed | 6 | 3 |
| Psychotic features at baseline, % | 58 | 60 |
| Assessment scale scores: mean (s.e.) | ||
| YMRS total | 37.5 (0.7) | 37.1 (0.7) |
| CGI severity | 4.1 (0.1) | 4.0 (0.1) |
| GAS | 34.6 (0.9) | 35.2 (0.9) |
| PANSS total | 54.2 (0.9) | 54.2 (0.9) |
| MADRS | 5.9 (0.4) | 5.1 (0.3) |
| Age at onset of bipolar disorder, years: mean (s.e.) | 25.2 (0.9) | 24.6 (0.7) |
| Previous manic episodes, n: mean (s.e.) | 4.8 (0.3) | 4.7 (0.4) |
Use of a pharmacological treatment for bipolar disorder during the 30 days before trial entry was reported by most patients (83% in the placebo group and 76% in the risperidone group). Psychotropic drugs had been taken by 83% of the placebo group and 75% of the risperidone group (including lorazepam by 42% and 43%, respectively, haloperidol by 41% and 36%, and chlorpromazine by 42% and 34%) and anti-epileptics by 17% of each group, carbamazepine being the most common (by 14% and 12%, respectively). During the wash-out period and the first 10 days of the double-blind treatment period, 99% of the patients received lorazepam. The mean daily dosage of lorazepam during the first 10 days of the double-blind period was 4.1 mg (s.d.=1.7) in the risperidone group and 4.4 mg (s.d.=1.6) in the placebo group (range 1–8 mg).
The trial was completed by 89% of patients in the risperidone group and 71% in the placebo group (Table 2). The most common reason for discontinuation was insufficient response (reported by 5% of risperidone patients and 15% of placebo patients). The rate of discontinuation owing to adverse events was similar in the risperidone and placebo groups (in 2% and 3% of patients, respectively). The mean modal dose of risperidone was 5.6 mg/day (s.e.=0.1).
Table 2 Trial completion rates and reasons for discontinuation

| Placebo group (n=144) % | Risperidone group (n=146) % | |
|---|---|---|
| Completed | 71 | 89 |
| Discontinued | 29 | 11 |
| Insufficient response | 15 | 5 |
| Lost to follow-up | 7 | 1 |
| Adverse event | 2 | 3 |
| Withdrew consent | 4 | 1 |
| Other | 1 | 1 |
Efficacy
All patients who received study medication and had at least one post-baseline assessment were included in the efficacy analysis. Substantial improvements were noted in patients given risperidone. Their mean YMRS total score was reduced from 37.1 (s.e.=0.7) at baseline to 14.5 (1.1) at end-point (P<0.001) (Fig. 2). The reductions in YMRS score at weeks 1–3 and at end-point were significantly greater among patients receiving risperidone than placebo. Reductions in YMRS scores were significantly greater in the risperidone group than in the placebo group at week 1 (mean change -11.7, s.e.=0.8 v. -8.3, s.e.=0.8; P<0.01), week 2 (mean change -20.2, s.d.=0.9 v. -11.4, s.e.=1.2; P<0.001), week 3 (mean change -25.2, s.e.=1.0 v. -15.0, s.e.=1.4; P<0.001) and at end-point (mean change -22.7, s.e.=1.1 v. -10.5, s.e.=1.3; P<0.001). In the primary between-treatment comparison, least-squares mean change in YMRS total score at end-point was -23.2 (s.d.=13.4) in the risperidone group and -10.8 (s.d.=13.4) in the placebo group (95% CI -15.6 to -9.3, P<0.001).
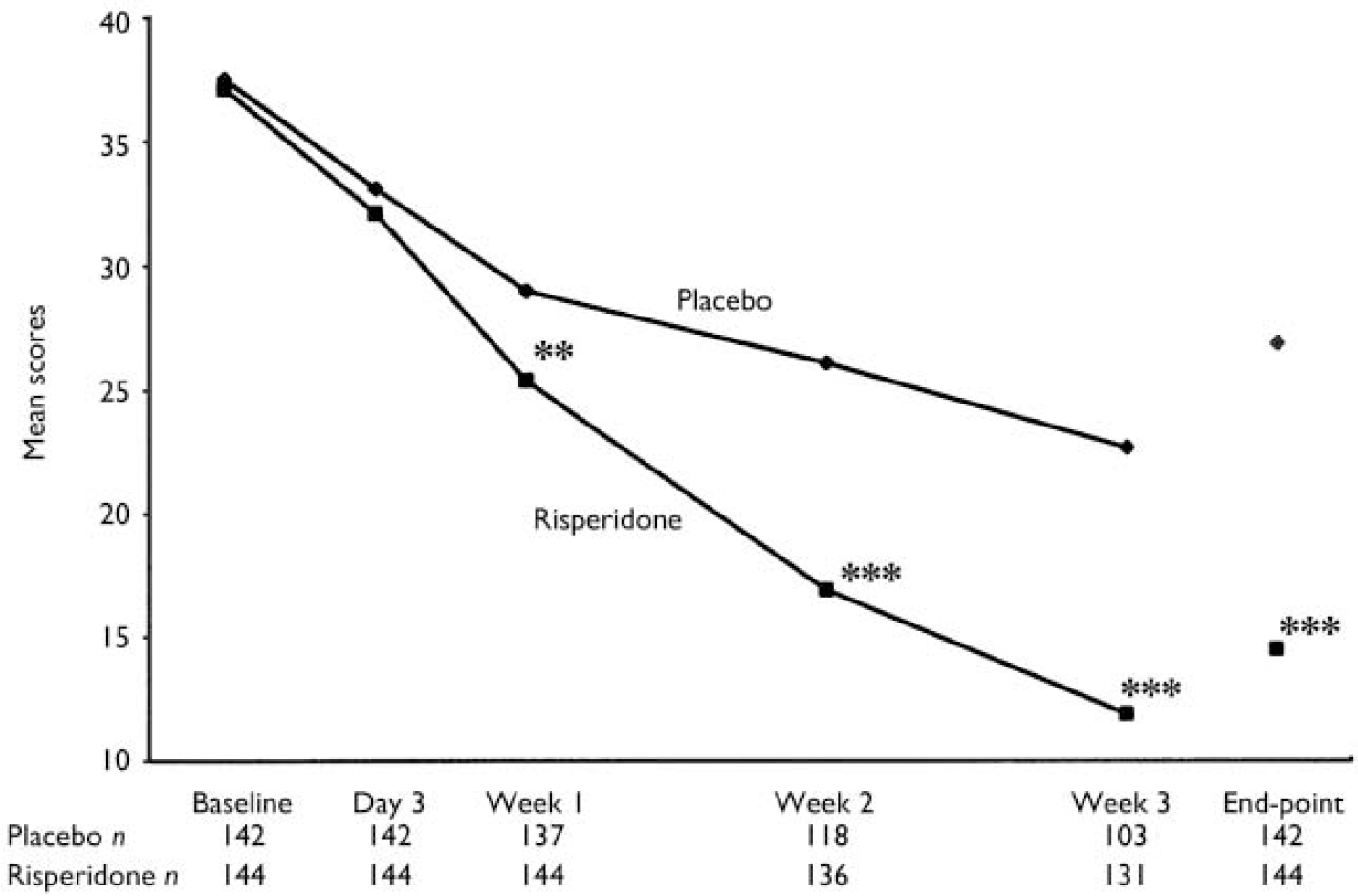
Fig. 2 Mean Young Mania Rating Scale scores in patients receiving placebo (♦) or risperidone (▪). Observed-case data are reported at day 3 and weeks 1–3 and last observation carried forward data at end-point (** P<0.01, *** P<0.001v. placebo).
At end-point, a clinical response (50% or greater decrease in YMRS score) was observed in 73% of patients given risperidone and 36% of patients given placebo (P<0.001). Compared with the placebo group, patients receiving risperidone demonstrated significantly greater improvements from baseline to end-point in mean CGI severity scores, from 4.0 (s.e.=0.1) to 2.0 (s.e.=0.1) in the risperidone group and from 4.1 (s.e.=0.1) to 3.2 (s.e.=0.1) in the placebo group (P<0.001) (Fig. 3). This was also the case for mean GAS scores, which increased from 35.0 (s.e.=0.9) at baseline to 62.7 (s.e.=1.4) at end-point in the risperidone group (P<0.001), and for PANSS total scores and scores on the positive symptoms and uncontrolled excitement/hostility factors (Fig. 4). Scores on the three other PANSS factors – negative symptoms, disorganised thoughts and anxiety/depression – were negligible at both baseline and end-point.
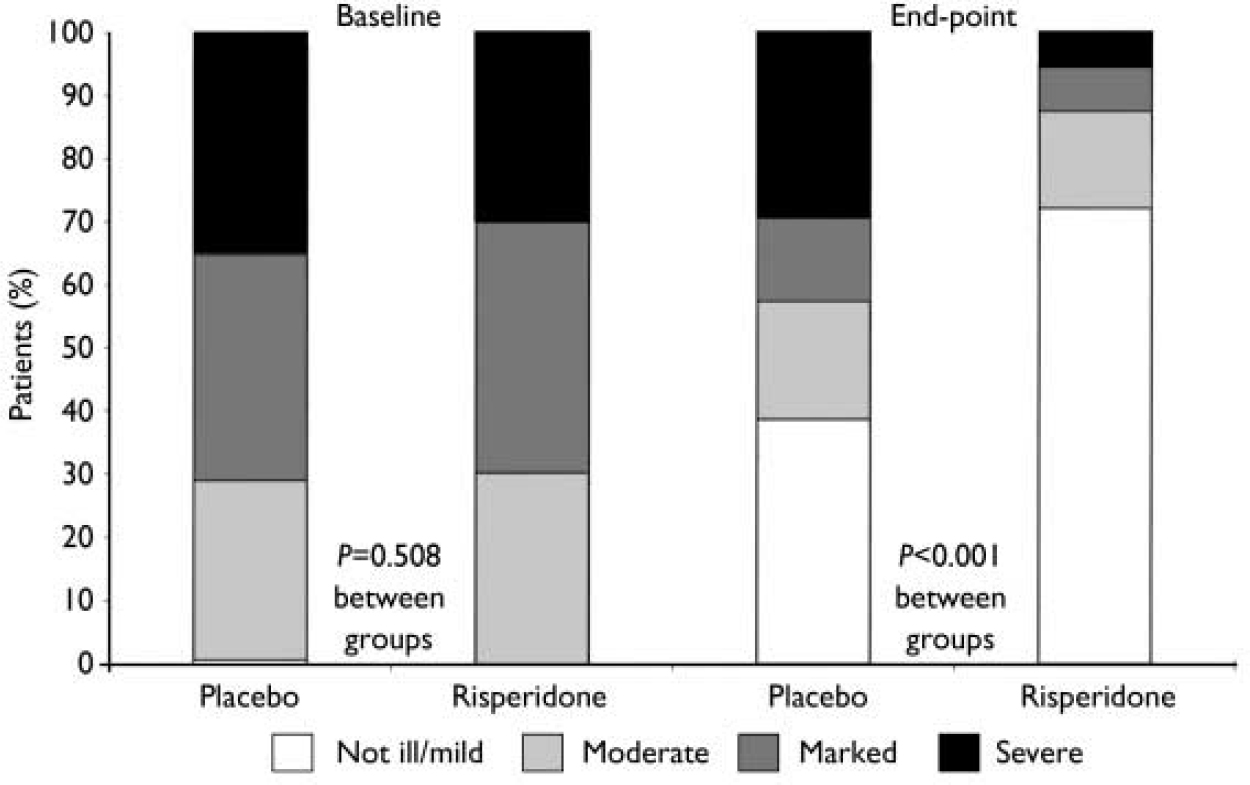
Fig. 3 Patients rated for illness severity on the Clinical Global Impression severity scale (not ill/mild, moderate, marked or severe/extremely severe) at baseline and end-point assessments.
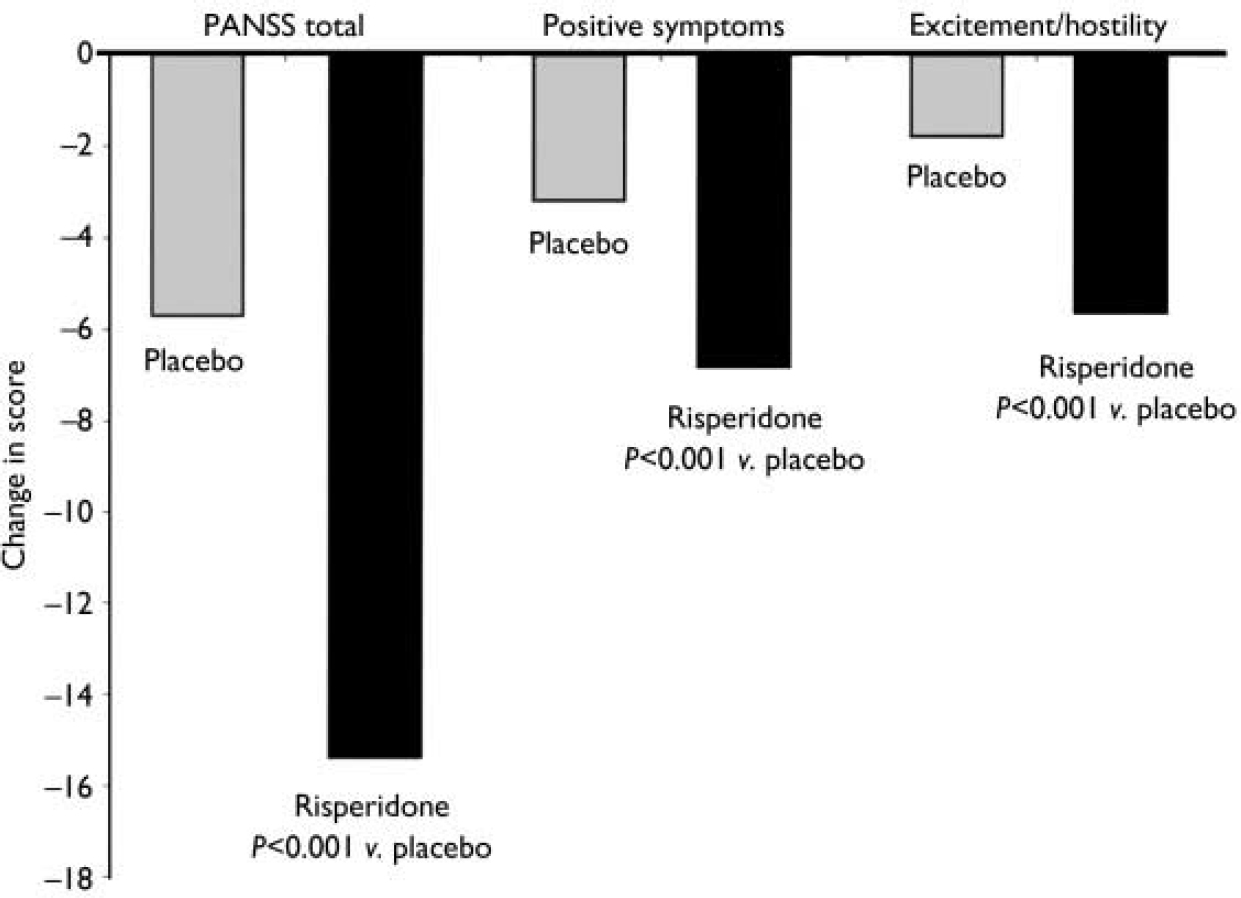
Fig. 4 Improvements in mean Positive and Negative Syndrome Scale (PANSS) total scores and scores on the positive symptoms sub-scale and uncontrolled hostility/excitement factors at end-point.
Improvement shown by reduction in MADRS score was significantly greater in the risperidone group than in the placebo group at day 3 (mean change -1.7, s.e.=0.2 v. -1.2, s.e.=0.2; P<0.01), week 1 (mean change -2.9, s.e.=0.2 v. -2.1, s.e.=0.3; P<0.01), week 2 (mean change -3.3, s.e.=0.3 v. -2.4, s.e.=0.3; P<0.001), week 3 (mean change -3.5, s.e.=0.5 v. -3.2, s.e.=0.4; P<0.001) and at end-point (mean change -3.2, s.e.=0.4 v. -2.5, s.e.=0.3; P<0.001). Baseline MADRS scores were low (5.9 in the placebo group and 5.1 in the risperidone group) in this patient population and 50–60% of the score was accounted for by the scale's reduced-sleep item.
Subgroup analyses
Among patients with or without psychotic features and those with a manic or mixed episode at baseline, patients who received risperidone demonstrated significantly greater improvements in YMRS score than those given placebo (Table 3). Results of analyses of patients by age group, gender and baseline YMRS score all indicated that risperidone was significantly superior to placebo in reducing symptoms, and no major difference in response was observed between these groups.
Table 3 Changes at end-point in Young Mania Rating Scale scores in patients with and without psychotic features and with a manic or mixed episode at baseline

| Placebo | Risperidone | 95% CI | |||
|---|---|---|---|---|---|
| n | Mean (s.e.) | n | Mean (s.e.) | ||
| Psychotic features | |||||
| Present | 81 | –10.2 (1.7) | 86 | –23.7 (1.4)*** | –18.6 to –10.4 |
| Absent | 61 | –11.0 (2.0) | 58 | –21.1 (1.9)*** | –14.4 to –4.4 |
| Episode | |||||
| Manic | 134 | –10.7 (1.3) | 141 | –22.5 (1.1)*** | –15.5 to –9.2 |
| Mixed | 8 | –7.9 (5.8) | 3 | –28.7 (11.8) | –69.8 to 15.8 |
Safety
Adverse events related to extrapyramidal symptoms included extrapyramidal disorder in 35% of the risperidone group and 6% of the placebo group, tremor in 10% and 1% respectively, dystonia in 5% and 0%, hyperkinesia in 1% and 0%, and oculogyric crisis in 1% and 0%. These adverse events were most often mild (59%) or moderate (33%) in the risperidone group. The only other adverse event observed in at least 10% of patients in either treatment group was insomnia (in 6% of the risperidone group and 10% of the placebo group). The median ESRS total scores (Parkinsonism plus dystonia plus dyskinesia) were 0 at both baseline and end-point in both treatment groups; median change scores were also 0 at all time points. In the risperidone group, for participants exhibiting a change in ESRS total scores, the change was generally between 1 and 5 on a scale ranging from 0 to 102. Antiparkinsonian medication was taken by 36% of the risperidone group and 6% of the placebo group.
Serious adverse events occurred infrequently and the rates of such events were similar in both treatment groups. Three patients in the placebo group and five in the risperidone group experienced adverse events that led to treatment discontinuation. No death occurred in this study. No clinically meaningful difference in vital signs or electrocardiographic data occurred between the two groups at any point during the study. The QTc interval did not exceed 500 ms in any patient in either group, regardless of the correction factor used in the analysis. Clinical laboratory values, other than that for prolactin, did not differ between the two treatment groups. Mean prolactin concentrations increased from baseline to end-point in the risperidone group (from 19 ng/ml to 86 ng/ml). No prolactin-related adverse event, however, was reported by any patient in either treatment group. Weight gains were comparable in the risperidone and placebo groups (0.07 kg in the placebo group and 0.06 kg in the risperidone group).
DISCUSSION
Patients given risperidone demonstrated significantly greater improvements than those given placebo on each of the efficacy measures (YMRS, CGI, GAS and PANSS). Improvements in YMRS total scores at end-point (the primary between-treatment comparison) were a reduction of 23.2 in the risperidone group and 10.8 in the placebo group, a between-treatment difference of 12.4 points. Significant between-treatment differences were seen as early as week 1 and were maintained during weeks 2 and 3 and at end-point. A clinical response (50% or greater reduction in YMRS score) at week 3 was noted in 73% of the patients taking risperidone and in 36% of the placebo group. Risperidone was equally efficacious in patients with and without psychotic features at baseline, indicating that its efficacy in controlling manic symptoms is independent of its antipsychotic effects.
Symptom severity
The patients with bipolar disorder who participated in the study were enrolled at several sites in India. Their symptoms of mania were substantially more severe than those of patients with bipolar disorder participating in trials elsewhere. The mean YMRS score at baseline was 37.0 in the risperidone group, in contrast to mean scores of 28.0 to 29.3 in controlled studies of risperidone and olanzapine conducted in the USA (Reference Tohen, Jacobs and GrundyTohen et al, 2000; Reference Sachs, Grossman and GhaemiSachs et al, 2002), in the six countries of an international study (Reference Yatham, Grossman and AugustynsYatham et al, 2003) and in South Africa (Reference Segal, Berk and BrookSegal et al, 1998). The Indian patients’ response to 3 weeks’ treatment with risperidone (a mean change of -23.2 in YMRS score) was unprecedented; for example, mean changes in YMRS score of -10.3 and -14.8 after 3 or 4 weeks’ treatment with olanzapine, -13.8 after 3 weeks’ treatment with quetiapine and -16.2, -14.5 and -14.3 after 3 or 4 weeks’ treatment with risperidone have been reported (Reference Segal, Berk and BrookSegal et al, 1998; Tohen et al, Reference Tohen, Sanger and McElroy1999, Reference Tohen, Jacobs and Grundy2000; Sachs et al, Reference Sachs, Grossman and Ghaemi2002, Reference Sachs, Chengappa and Suppes2004; Reference Yatham, Grossman and AugustynsYatham et al, 2003).
Baseline scores on the MADRS were generally low in this population and mostly were accounted for by the reduced-sleep item on that scale. The significant reduction in MADRS score from baseline to end-point that was observed with risperidone was primarily due to an improvement in scores on the reduced-sleep item. The improvement in sleep might have been an epiphenomenon of the improvement of manic symptoms with risperidone. The overall reduction in MADRS scores is reassuring and indicates no induction of depression with risperidone.
Adverse events
Treatment with risperidone was well tolerated. The 3-week trial was completed by 89% of patients randomly assigned to risperidone. Extrapyramidal symptoms were reported more frequently by patients taking risperidone than by those taking placebo and more patients in the risperidone group (36%) than in the placebo group (6%) received antiparkinsonian medications. The severity of these movement disorders, however, was generally mild: median ESRS total scores were 0 at both baseline and end-point in both groups. Only one patient discontinued the trial because of movement disorders, suggesting that the disorders were not perceived as severe and that symptoms, if present, responded adequately to treatment with antiparkinsonian medication. The movement disorders reported by these patients may be related to the high baseline YMRS scores and the resultant rapid increase in risperidone dosage. The patients’ low body weight (mean 54 kg), together with a mean modal dose of 5.6 mg per day of risperidone, might also have contributed to these adverse events. Patients with particularly severe manic symptoms may need dosages of risperidone as high as 0.10 mg/kg. In clinical practice this need for a higher dosage needs to be balanced by the potentially increased risk of movement disorders.
Except for a change in prolactin concentration, no clinically meaningful change was observed in mean laboratory values from baseline to end-point. No prolactin-related adverse event was reported. Neither group demonstrated any clinically significant change in vital signs, body weight or electrocardiograms (including QTc prolongation). According to adverse events reports, there was no evidence of mania exacerbation among patients given risperidone.
Risperidone was generally well tolerated, as evidenced by the low incidence of other adverse events and the high completion rate.
Trial completion
The study was completed by 89% of the patients in the risperidone group and 71% in the placebo group. This percentage is high compared with other placebo-controlled trials of similar duration in patients with bipolar mania. Several reasons for this can be suggested: for example, the patients in this trial were more severely ill than those usually included in placebo-controlled trials, and many remained in hospital for the entire trial period, which was not routinely the case in other trials. Both of these factors, in addition to a healthcare environment different from that in Europe or the USA, might have positively influenced retention rates. Although our trial lasted only 3 weeks, the data are similar to those from a 6-month naturalistic study of risperidone in bipolar mania (Reference Vieta, Goikolea and CorbellaVieta et al, 2001): in that study, 83% of patients completed the 6-month period, and their mean baseline YMRS total score of 29.2 was reduced to 11.1 at week 4, with further reduction to a mean score of 2.8 at 6 months. The similarity of the results observed in our trial and in the naturalistic study raises the question of whether the longer-term results of the naturalistic study could be predictive for controlled clinical trials.
Early and substantial patient response
In summary, patients with bipolar disorder and severe manic symptoms who received risperidone monotherapy improved on all measures of efficacy. Treatment with risperidone produced significant improvement as early as week 1 and substantial improvements in YMRS score at end-point, when there was a 12-point difference between risperidone and placebo mean scores. Despite the aggressive dosing regimen in this trial, risperidone was well tolerated: more patients who received the drug (89%) than those who received placebo (71%) completed the study. The results confirm those of other trials involving diverse patient populations (Reference Ghaemi, Sachs and BaldassanoGhaemi et al, 1997; Reference Segal, Berk and BrookSegal et al, 1998; Vieta et al, Reference Vieta, Goikolea and Corbella2001, Reference Vieta, Brugue and Goikolea2004; Reference Sachs, Grossman and GhaemiSachs et al, 2002; Reference Yatham, Grossman and AugustynsYatham et al, 2003), in which risperidone was found to be effective and safe in patients with acute mania.
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
▪ Risperidone was associated with relief of mania: significant improvement was observed as early as the first and second weeks after therapy began and, by the third week, 79% of patients receiving the drug demonstrated a therapeutic response.
-
▪ Risperidone was equally efficacious in patients with or without psychotic features at baseline, indicating that its efficacy in controlling manic symptoms is independent of its antipsychotic effects.
-
▪ Despite the aggressive treatment, risperidone was well tolerated: more patients who received the drug completed the study than those who received placebo.
LIMITATIONS
-
▪ Although mood stabilisers were not administered concomitantly with risperidone, most patients did receive lorazepam.
-
▪ The healthcare environment in which the trial was conducted may limit generalisations of its results to other settings.
-
▪ At the doses employed risperidone induced extrapyramidal side-effects that may be dose-related and should be carefully monitored in the clinical situation.








eLetters
No eLetters have been published for this article.