


Serotonergic (5-HT) neurotransmission contributes to many physiological functions, including appetite, sleep, aspects of cognition and the expression of depression and anxiety. The 5-HT transporter (5-HTT) is central to the regulation of brain and peripheral serotonergic neurotransmission. A functional polymorphism in the promoter region of the serotonin transporter gene (SLC6A4), known as 5-HTTLPR, has been associated with depression and with risk factors for depression (e.g. neuroticism), with the short ‘s’ allele reducing the transcriptional efficiency of the serotonin promoter and leading to decreased 5-HTT expression (Reference Lesch, Bengel and HeilsLesch et al, 1996). Although a recent meta-analysis did not support a direct relationship between the 5-HTTLPR and depression onset (Reference Anguelova, Benkelfat and TureckiAnguelova et al, 2003), the ‘s’ allele was thought to increase vulnerability to adverse life events.
A longitudinal cohort study (Reference Caspi, Sugden and MoffittCaspi et al, 2003) which tested this gene × environment interaction hypothesis reported that participants with the s/s genotype were more likely than those with the s/l or l/l genotypes to experience DSM major depression in the presence of increasing numbers of adverse life events.
We sought to replicate a gene × environment interaction in a cohort with individualised longitudinal data for life events and onset of major depression. We also examined data for a broad range of experience of depression and putative risk factors for depression, including parental bonding and neuroticism.
METHOD
Study protocol
The cohort of 165 young adults (initially consisting of 109 women and 56 men; mean age=23.4 years, s.d.=4.3 years) was recruited in 1978 from a group undertaking a 1-year postgraduate teacher training course (at a Sydney Teachers College) to take part in a longitudinal study designed to investigate gender differences in rates of anxiety and depression. The cohort has been followed up at 5-yearly intervals (Reference Wilhelm and ParkerWilhelm & Parker, 1989). At each follow-up (in 1983, 1988, 1993 and 1998) all of the participants completed a series of self-report questionnaires in conjunction with a semi-structured interview asking about work and personal life events for each year over the 5-year period, as well as about alcohol and substance misuse, medical history and methods of coping with stress, depression and adversity. The questionnaires measured neuroticism (Reference Eysenck and EysenckEysenck & Eysenck, 1964), ‘normal depression’ (mood fluctuation and symptoms consistent with major depression or dysthymia, but only lasting for minutes to days) (Reference Parker, Wilhelm and AsghariParker et al, 1998; Reference Wilhelm, Parker and AsghariWilhelm et al, 1998), ‘trait depression’ (a person's tendency to experience a depressive mood) (Reference Costello and ComreyCostello & Comrey, 1967) and parental bonding. The latter was assessed with the Parental Bonding Instrument (PBI), which consists of sub-scales that measure care and overprotection for each parent up to the age of 16 years (Reference Parker, Tupling and BrownParker et al, 1979). The PBI was completed at the first three follow-up intervals and then again in 1998.
The age at onset and the frequency of major depression and anxiety disorders were also recorded at all follow-up intervals, using the Diagnostic Interval Schedule (DIS; Reference Robins, Helzer and CroughanRobins et al, 1981) in 1983 and 1988, and the Composite International Diagnostic Interview (CIDI; Reference Robins, Helzer and BurrowsRobins & Helzer, 1988) in 1993, 1998 and 2003. A diagnosis of lifetime major depression was made by employing an ‘add-on’ strategy in which the lifetime history was compiled by adding on new episodes identified at each wave of follow-up, respecting the decision about the presence or absence of episodes that was made at the interview closest to the index episode (Reference Wilhelm and ParkerWilhelm & Parker, 1994). At each wave of follow-up, any anxiety or depressive disorders that were identified were recorded on a life chart, which also documented the occurrence of any of a range of significant life events. Both positive events (e.g. having a child or being promoted) and adverse events (e.g. marital breakdown or death of a parent) were recorded. During the period 2002–2003, participants were shown their individual life charts, which included all of the material that had been collected during the course of the study. The aim was to verify the history of depression and/or anxiety disorders, the treatment history and the presence and type of positive and adverse life events pertaining to work, health, friends and family on the basis of data collected on a year-by-year basis at each 5-yearly interview. These charts were viewed before any consideration of the genetic study reported here.
By 2003, 149 of the original 165 participants remained in the study (8 had died, 2 could not be located, 2 were too ill to continue and 4 did not wish to be further involved in the study). Of these 149 individuals, 62 (42%) met the criteria for lifetime major depression, with a mean age at onset of 30.7 years (s.d.=8.2, range=15–50 years). In total, 128 members of the cohort gave their informed consent for the collection of genetic material. Of the DNA samples, 91% (n=117) were obtained via blood and 9% were obtained via buccal swabs (for those individuals who did not wish to undergo phlebotomy). The genomic DNA was obtained and genotypes for the 5-HTT gene-linked promoter region were determined for 127 members of the cohort.
DNA extraction and genetic analysis
Genomic DNA was extracted from whole blood using a standard salting-out method (Reference Miller, Dykes and PoleskyMiller et al, 1988). To extract genomic DNA from cheek swabs, buccal cells were pelleted by centrifugation and digested overnight at 42°C in a 420-μl volume containing 1 mg/ml proteinase K, 6 mmol/l TRIS-Cl pH 7.5, 6 mmol/l ethylenediamine tetra-acetic acid (EDTA), 3% sodium sarkosyl, 1.2 mol/l guanidine hydrochloride and 0.5 mol/l ammonium acetate. DNA was purified from the digest by standard chloroform extraction and ethanol precipitation, and was resuspended in distilled water. The 5-HTTLPR was amplified with the following primers: forward 5′-TGCCGCTCTGAATGCCAG CAC-3′, and reverse 5′-GCGGGATTCTG GTGCCACCTA-3′, to generate a 464-base pair (bp) product for the 16-repeat (l) allele, and a 420-bp product for the 14-repeat (s) allele. The polymerase chain reaction (PCR) was performed in 25-μl volumes containing Optiprime mastermix (Stratagene), Optibuffer I (Stratagene), 400 mmol/l betaine, 160 mmol/l 7-deaza-2”deoxyguanosine 5”-triphosphate, 20 ng template DNA, 20 pmol for each primer, and 1 U of AmpliTaq Gold DNA polymerase (Applied Biosystems). Reactions included initial denaturation at 94°C for 12 min, followed by 35 cycles at 94°C for 30 s, 65°C for 45 s, 72°C for 1 min, and a final extension of 10 min at 72°C. After electrophoresis in 2% agarose gels, products were visualised under ultraviolet light.
Statistical methods
Logistic regression analysis was used to assess the association between depression and the 5-HTTLPR genotype, adverse life events and the gene × environment interaction, with gender included as a covariate. As the age of cohort members was controlled for in the original sample selection procedures (Reference Wilhelm and ParkerWilhelm & Parker, 1989), it was not included as a covariate in the present analysis (depression= β0+β1(gender)+β2 (5-HTTLPR)+β3 (number of adverse life events)+β4 (5-HTTLPR × number of adverse life events)).
In the above model, β0 is the intercept, β1 is the gender coefficient (0=female, 1=male), β2 is the 5-HTTLPR coefficient (0=‘s/s’, 1=‘s/l’, 2=‘l/l’), β3 is the coefficient for adverse life events (0=none, 1=1, 2=2, 3=3 or more) and β4 is the interaction-effect coefficient, which is the product of the two variables (5-HTTLPR and adverse life events). Life events were examined over two time periods, namely 1 year and 5 years before the onset of depression. For participants who had no history of depression, the number of life events was calculated for the 1- and 5-year periods before the mean age of the first episode of major depression (30.6 years) in the sample of people with depression. Adverse and then positive life events were examined in separate regression models. Analyses of variance (ANOVAs) were used to analyse associations between the 5-HTTLPR genotype and the following: self-reported vulnerability factors (neuroticism, ‘normal’ and ‘trait’ depression, parental bonding); total number of lifetime adverse life events; and total number of positive life events. Genotype variations were treated as the independent variable. The dependent variables were baseline scores on the self-report measures (each scale being independently analysed), adverse life events and positive life events (each summed across the participant's lifetime).
RESULTS
The 127 cohort members had a mean age (in 2003) of 47.7 years (s.d.=2.8), and consisted of 85 women and 42 men. They were divided into three groups on the basis of their 5-HTTLPR genotype (s/s, s/l or l/l). The allele frequencies were 0.457 (s) and 0.543 (l), and the genotype frequencies (s/s, n=27, 0.21; s/l, n=62, 0.49; l/l, n=38, 0.30) were in Hardy–Weinberg equilibrium (χ2=0.03 (2), P=0.98). There was no gender difference in genotype frequencies (χ2=3.56 (2), P=0.17).
Of the 127 cohort members, 53 (42%) met the criteria for lifetime major depression, with a mean age at onset of 30.6 years (s.d.=8.4, range=15–50 years). Of those who had experienced at least one episode of major depression, 32% reported no adverse life events, 15% reported one, 28% reported two and 25% reported three or more adverse life events in the 5 years before their first episode of major depression. Of those with no history of major depression, 32% reported no adverse life events, 34% reported one, 22% reported two and 12% reported three or more adverse life events in the same period.
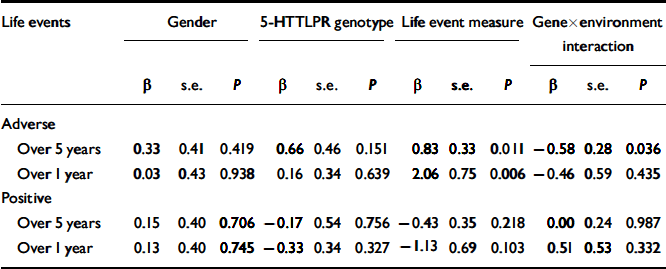
The results of the logistic regression analyses are shown in Table 1. When adverse life events were examined, a main effect of the 5-HTTLPR was not found. However, an interaction between the 5-HTTLPR and adverse life events was found to significantly predict the onset of major depression for the 5 years before depression onset. Specifically, the influence of adverse life events on the onset of major depression was significantly greater for individuals with the s/s genotype (Fig. 1). This interaction was not significant for the single year before depression onset (Fig. 2). Overall, adverse life events were found to be the strongest predictor of major depression.
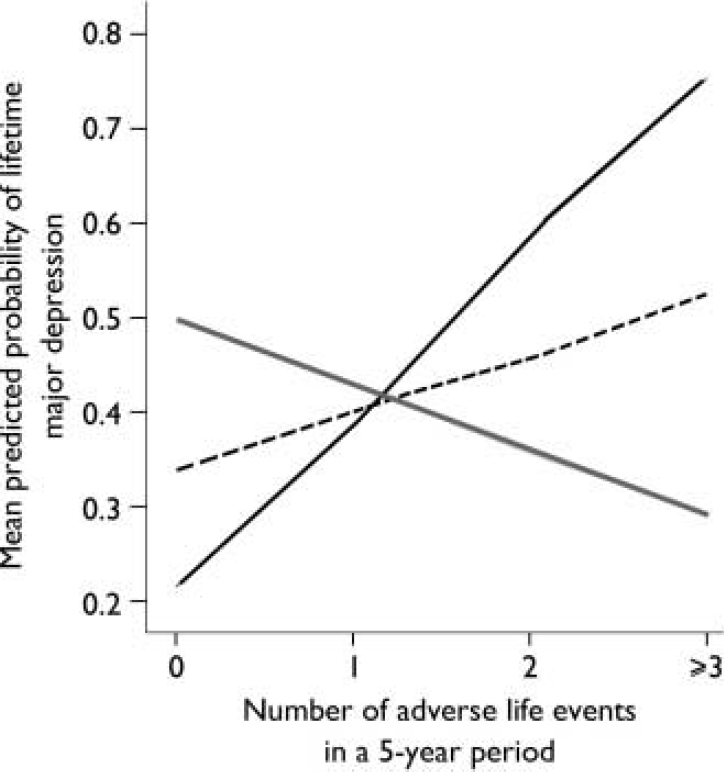
Fig. 1 Effect of number of adverse life events during a 5-year period on mean predicted probability of major depression.—, s/s; - - - -, s/l; ![]() , l/l.
, l/l.
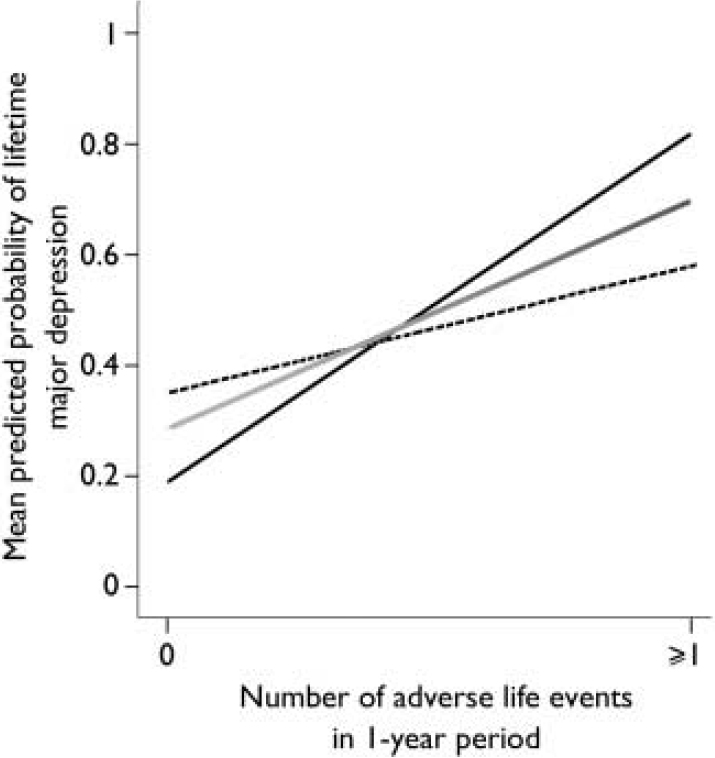
Fig. 2 Effect of number of adverse life events during a 1-year period on mean predicted probability of major depression.—, s/s; - - - -, s/l; ![]() , l/l.
, l/l.
Table 1. Binary logistic regressions of gender, genotype, life events and gene × environment interaction on lifetime history of major depression for the 127 cohort members
| Life events | Gender | 5-HTTLPR genotype | Life event measure | Gene × environment interaction | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| β | s.e. | P | β | s.e. | P | β | s.e. | P | β | s.e. | P | |
| Adverse | ||||||||||||
| Over 5 years | 0.33 | 0.41 | 0.419 | 0.66 | 0.46 | 0.151 | 0.83 | 0.33 | 0.011 | -0.58 | 0.28 | 0.036 |
| Over 1 year | 0.03 | 0.43 | 0.938 | 0.16 | 0.34 | 0.639 | 2.06 | 0.75 | 0.006 | -0.46 | 0.59 | 0.435 |
| Positive | ||||||||||||
| Over 5 years | 0.15 | 0.40 | 0.706 | -0.17 | 0.54 | 0.756 | -0.43 | 0.35 | 0.218 | 0.00 | 0.24 | 0.987 |
| Over 1 year | 0.13 | 0.40 | 0.745 | -0.33 | 0.34 | 0.327 | -1.13 | 0.69 | 0.103 | 0.51 | 0.53 | 0.332 |
No significant differences were found when positive life events were examined in the equivalent time periods preceding the onset of major depression. Neither the 5-HTTLPR, positive life events nor their interaction predicted depression onset. Gender was not a significant covariate in the model for any of the regression analyses.
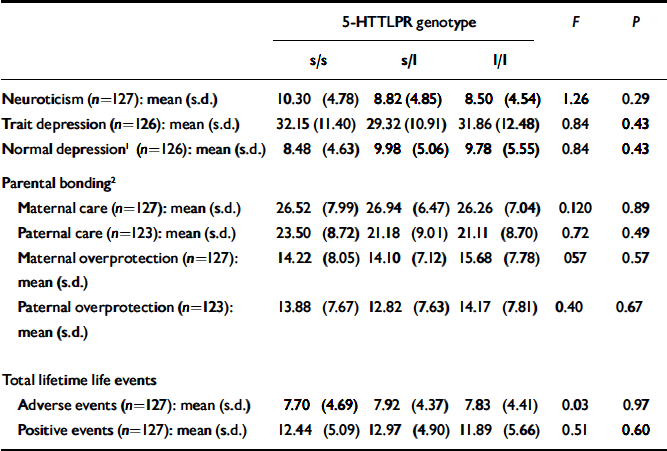
Table 2 lists the mean scores for neuroticism, normal and trait depression measures, parental bonding, and lifetime adverse and positive life events. No significant differences were found when data were analysed according to genotype.
Table 2. Baseline self-report measures and total lifetime life events according to 5-HTTLPR genotype
| 5-HTTLPR genotype | F | P | |||
|---|---|---|---|---|---|
| s/s | s/l | l/l | |||
| Neuroticism (n=127): mean (s.d.) | 10.30 (4.78) | 8.82 (4.85) | 8.50 (4.54) | 1.26 | 0.29 |
| Trait depression (n=126): mean (s.d.) | 32.15 (11.40) | 29.32 (10.91) | 31.86 (12.48) | 0.84 | 0.43 |
| Normal depression1 (n=126): mean (s.d.) | 8.48 (4.63) | 9.98 (5.06) | 9.78 (5.55) | 0.84 | 0.43 |
| Parental bonding2 | |||||
| Maternal care (n=127): mean (s.d.) | 26.52 (7.99) | 26.94 (6.47) | 26.26 (7.04) | 0.120 | 0.89 |
| Paternal care (n=123): mean (s.d.) | 23.50 (8.72) | 21.18 (9.01) | 21.11 (8.70) | 0.72 | 0.49 |
| Maternal overprotection (n=127): mean (s.d.) | 14.22 (8.05) | 14.10 (7.12) | 15.68 (7.78) | 057 | 0.57 |
| Paternal overprotection (n=123): mean (s.d.) | 13.88 (7.67) | 12.82 (7.63) | 14.17 (7.81) | 0.40 | 0.67 |
| Total lifetime life events | |||||
| Adverse events (n=127): mean (s.d.) | 7.70 (4.69) | 7.92 (4.37) | 7.83 (4.41) | 0.03 | 0.97 |
| Positive events (n=127): mean (s.d.) | 12.44 (5.09) | 12.97 (4.90) | 11.89 (5.66) | 0.51 | 0.60 |
DISCUSSION
The present study has successfully replicated the findings of Caspi et al (Reference Caspi, Sugden and Moffitt2003) in demonstrating a significant interaction between the 5-HTTPLR genotype and adverse events in determining the likelihood of an episode of major depression in a longitudinal cohort. Whereas Caspi et al (Reference Caspi, Sugden and Moffitt2003) looked for determinants of depression occurring between the ages of 21 and 26 years, our cohort members had individual lifetime charts from data that had been collected previously, and which recorded positive and adverse life events, age at onset and number of episodes of clinical depression and anxiety disorders. This allowed us to determine the lifetime experience of depression in a sample for which longitudinal data on the occurrence of life events and age at onset of major depression were available for each individual.
Characteristics of the sample
Our study utilised a relatively small group of individuals who were homogeneous with regard to a number of socio-demographic variables (age, socio-economic status, education and ethnicity) (Reference Wilhelm and ParkerWilhelm & Parker, 1989) compared with the larger sample of 847 participants in the Dunedin birth cohort that was investigated by Caspi et al (Reference Caspi, Sugden and Moffitt2003). All members of the group had received tertiary education, and we have previously reported low rates of cigarette and alcohol consumption and a negligible forensic history in this group (Reference Wilhelm and ParkerWilhelm & Parker, 1993; Reference Wilhelm, Parker and Hadzi-PavlovicWilhelm et al, 1997). The total cohort and the smaller group which provided genetic material have identical and relatively high rates of major depression (42%). We have previously argued that these high rates are related to the following: the serial data collection method that was employed, which captures single episodes or less severe episodes that had not attracted clinical services, or which could have been overlooked or forgotten later; and the lack of antisocial behaviour, substance misuse and dependence that may serve as ‘depressive equivalents’ in other samples (Wilhelm & Parker, Reference Wilhelm and Parker1989, Reference Wilhelm and Parker1993, Reference Wilhelm and Parker1994; Reference Wilhelm, Parker and Hadzi-PavlovicWilhelm et al, 1997). The argument that longitudinal studies with multiple points of data collection yield higher rates of recall of episodes is reinforced by the findings of another longitudinal study (Reference Wells and HorwoodWells & Horwood, 2004), which reported rates of 37% for DSM major depression among young people assessed at yearly intervals from the age of 14 to 21 years. However, at 25 years of age, the same participants commonly failed to recall episodes and key symptoms of lifetime major depression. Similarly, 40% of respondents who reported a lifetime history of depressive episodes at baseline in another longitudinal survey (Reference Thompson, Bogner and CoyneThompson et al, 2004) failed to do so at a 13-year follow-up.
Life events and depression onset
The association between adverse life events and onset of major depression is well established in the literature (Reference Kendler, Karkowski and PrescottKendler et al, 1999). In our study, variations in the 5-HTTLPR did not independently predict the onset of major depression, but its interaction with adverse life events was significant for the 5-year period. Although Caspi and colleagues did not report on the relationship between the 5-HTTLPR genotype and the number of life events experienced within the 12-month period before depression onset, we also examined this time period as being potentially most directly related to the onset of major depression. We found a strong main effect, suggesting that the recent experience (within the last 12 months) of adverse life events predicts depression onset. However, we found no evidence of a significant interaction between the 5-HTTLPR genotype and adverse life events. This may be because of the time frame, which did not allow for a sufficient range of life events. Therefore, we used a single dichotomous distinction (i.e. no adverse life events v. one or more adverse life events) between cohort members’ experiences within the 12-month period. The small sample size also limited the statistical power, and in this respect a larger sample might reveal the expected gene × environment interaction for the shorter time period leading up to the first depressive episode. As positive life events may increase emotional responsiveness, we also examined the effect of positive life events on onset of major depression. Positive life events were not found to exert a depressogenic effect, and instead we found a trend in the opposite direction. In a larger sample, positive life events may exert a protective effect.
Genetic and psychological risk factors for major depression
About 50% of the variation in neuroticism has been attributed to genetics (Reference LoehlinLoehlin, 1992). Neuroticism is the most consistent risk factor for the onset and relapse of major depression (Reference Eccleston and ScottEccleston & Scott, 1991; Reference Wilhelm, Parker and Dewhurst-SavellisWilhelm et al, 1999), and has been shown to interact with adverse environmental factors in the aetiology of major depression, such that higher neuroticism infers greater sensitivity to the depressogenic effects of adverse life events (Reference Kendler, Kuhn and PrescottKendler et al, 2004). Yet the search for candidate genes has yielded conflicting results (Reference Schinka, Busch and Robichaux-KeeneSchinka et al, 2004). A polymorphic variation in the 5-HTTLPR was found to be associated with anxiety-related personality traits (Reference Lesch, Bengel and HeilsLesch et al, 1996). However, subsequent studies have come to conflicting conclusions (Reference Jorm, Henderson and JacombJorm et al, 1998; Reference Deary, Battersby and WhitemanDeary et al, 1999; Reference Greenberg, Li and LucasGreenberg et al, 2000).
Consistent with previous reports (e.g. Reference Lesch, Bengel and HeilsLesch et al, 1996), we postulated that individuals with the s/s genotype are more temperamentally sensitive, and as a result may report more life events, higher neuroticism as well as increased ‘trait’ and ‘normal’ depression scores, and a higher level of parental overprotection. We have previously reported that cohort members who had experienced repeated episodes of major depression had lower scores for maternal and paternal care and higher scores for trait depression and maternal overprotection recorded at baseline in 1978 (Reference Wilhelm, Parker and Dewhurst-SavellisWilhelm et al, 1999). We have also reported that women have consistently higher scores than men for neuroticism on the Eysenck Personality Inventory (EPI) (Reference Wilhelm, Parker and Dewhurst-SavellisWilhelm et al, 1999) and ‘normal depression’ (Reference Wilhelm, Parker and AsghariWilhelm et al, 1998), both of which have been associated with higher rates of major depression (Reference Parker, Wilhelm and AsghariParker et al, 1998; Reference Wilhelm, Parker and Dewhurst-SavellisWilhelm et al, 1999). However, none of these factors appeared to be associated with the 5-HTTLPR genotype. There was also no evidence of differential reporting of positive or adverse life events by individuals with the s/s, s/l or l/l genotypes.
Although individuals with the s/s genotype have a greater probability of experiencing onset of depression with increasing numbers of adverse life events, the trend for those with the l/l genotype is, if anything, in the opposite direction (Fig. 1). The same trend is noticeable in the earlier study (Reference Caspi, Sugden and MoffittCaspi et al, 2003), and may be a chance finding. There is no evidence that people with the l/l genotype report fewer adverse life events, and we speculate that they may be less reactive to environmental cues but possibly employ coping styles that modulate increasing levels of stress. Although interest has been focused on the short allele, it would be beneficial also to consider the stress-related experience of people with the long allele.
Previous attempts to replicate earlier findings
Four recent studies have also aimed to replicate the results obtained by Caspi and colleagues. First, in a negative replication, Gillespie et al (Reference Gillespie, Whitfield and Williams2004) cited factors such as the wide age range (19–78 years) of their sample and the measurement of life events at a single time point for a 12-month period as possible factors contributing to a reduction in power to detect interactions between life events and onset of major depression. For these reasons, too, their methodology was less likely to have captured the first onset of major depression than the method that we used.
In the second study, which utilised an adolescent sample, Eley et al, (Reference Eley, Sugden and Corsico2004) used a depression symptom measure as the outcome variable, and reported an interaction between the 5-HTTLPR and a composite measure of environmental risk that reached the level of significance for female but not for male participants. In the third study, Grabe et al (Reference Grabe, Lange and Wolff2005) analysed the genotype by grouping individuals with s/s and s/l genotypes together, and comparing their findings for these individuals with those for the l/l genotype. They found an interaction between the 5-HTTLPR and life stressors (unemployment and number of chronic diseases). However, this also only reached the level of significance in women.
These two partial replications (Reference Eley, Sugden and CorsicoEley et al, 2004; Reference Grabe, Lange and WolffGrabe et al, 2005) adopted very different methodologies to our study, as both were cross-sectional and used symptom rating scales as outcome measures. Both studies also used different environmental stressors. Rather than summing reported life events, they used either a composite of various stressors (Reference Eley, Sugden and CorsicoEley et al, 2004) or unemployment and number of diseases (Reference Grabe, Lange and WolffGrabe et al, 2005). These may constitute ‘chronic stressors’ which could reduce the impact of the postulated interaction and may not be directly comparable with the more acute adverse events that were used by Caspi's group (Reference Caspi, Sugden and MoffittCaspi et al, 2003) and in our study. Furthermore, cross-sectional cross-sectional designs do not allow clear identification of a causal pathway between life events and depression onset. Our use of life charts and a longitudinal design allowed more accurate identification of the onset of first episodes and preceding life events for each individual.
In a positive replication, Kaufman et al (Reference Kaufman, Yang and Douglas-Palumberi2004) studied children with a history of maltreatment and found that those with the s/s allele reported significantly more depressive symptoms than those with the s/l or l/l allele. There were no differences in the number of depressive symptoms between genotypes among controls (without a history of maltreatment). This study used depressive symptoms rather than major depression, and draws attention to the moderating role of social support in ‘at-risk’ individuals.
Gillespie et al (Reference Gillespie, Whitfield and Williams2004) noted the problems of looking for a gene × environment interaction in an older population, but there may also be problems with an adolescent population such as that used by Eley et al (Reference Eley, Sugden and Corsico2004). The mean age of depression onset for both men and women is in the midto late twenties (Reference Weissman, Bland and CaninoWeissman et al, 1996), and gender differences in rates start to emerge between the ages of 15 and 18 years (Reference Hankin, Abramson and MoffittHankin et al, 1998). Thus a sample of adolescents with an age range of 12–19 years may prematurely demarcate the group with depression, some of whom may not as yet have ‘declared themselves’. This may account both for the non-significant trend for the group as a whole (Reference Eley, Sugden and CorsicoEley et al, 2004) and for the significant gene × environment interaction in female participants. The findings of Kaufman et al (Reference Kaufman, Yang and Douglas-Palumberi2004) suggest that the gene × environment interaction is apparent even in childhood and adolescence when experience of environmental adversity varies considerably within the sample.
Although the positive studies (Reference Caspi, Sugden and MoffittCaspi et al, 2003; Reference Eley, Sugden and CorsicoEley et al, 2004; Reference Kaufman, Yang and Douglas-PalumberiKaufman et al, 2004; Reference Grabe, Lange and WolffGrabe et al, 2005) differ from the present one in methodology, they are consistent with it in that they all demonstrate that the 5-HTTLPR genotype is a significant factor determining the likelihood of experiencing depression after exposure to multiple adverse life events.
Implications
Both proximal factors (e.g. adverse life events) and distal factors (e.g. dysfunctional parenting, childhood maltreatment) can be involved in any putative gene × environment interaction in the onset of depression (Reference Farmer, Eley and McGuffinFarmer et al, 2005). The consistency of the findings of the present study with regard to proximal events raises the possibility of preventive interventions that would have potentially major clinical significance. The gene × environment interaction reported in this paper is modest, and it would be premature to apply this finding to the clinical arena, but it is none the less vital to seriously consider the ethical and policy ramifications of such genetic research (Reference Farmer and OwenFarmer & Owen, 1996; Reference Rutter and PlominRutter & Plomin, 1997; Reference Morley, Hall and CarterMorley et al, 2004). Future research efforts may be directed towards determining whether the identification of ‘at-risk’ genotypes can inform treatment decisions concerning psychological or pharmacological therapies to prevent or ameliorate the eventual onset of depression. In addition, there is the equally important question of what can be learned from studying ‘at-risk’ individuals who are resilient to the effects of adverse life events (Reference Farmer, Eley and McGuffinFarmer et al, 2005).
Clinical Implications and Limitations
CLINICAL IMPLICATIONS
-
▪ Some individuals seem to be genetically predisposed to the onset of major depression when confronted with a series of adverse life events.
-
▪ Further research is required to determine whether different coping strategies are more effective for patient groups with the different alleles.
-
▪ There may be a future role for preventive psychological and/or pharmacological interventions to help ‘at-risk’ patients to cope effectively with change and adversity.
LIMITATIONS
-
▪ The findings require further confirmation in a variety of clinical and non-clinical settings.
-
▪ It is unlikely that a single gene × environment interaction could explain vulnerability to the onset of depression.
-
▪ Ethical issues will need to be considered if patients ask to know whether they are genetically vulnerable to depression.
Acknowledgements
We thank the cohort members for their time and continuing interest, and for generously donating their samples for genotype analysis. We are also grateful to Emma Burgess for assistance with collection of blood samples, and Dusan Hadzi-Pavlovic for statistical advice.
This study was supported by NH & MRC Program Grants 222708, 230802 and 157209 and a New South Wales Centre for Health Infrastructure Grant.





eLetters
No eLetters have been published for this article.