


In this month's Journal McClellan et al contrast two models of the genetic architecture of schizophrenia. Here we provide a context for that paper by considering more widely the genetic and phenotypic complexity of psychosis and how this has an impact on genetic research.
COMPLEX GENETIC DISEASES
The term ‘complex genetic diseases’ refers to common familial illnesses that do not show a simple Mendelian pattern of inheritance (Reference Lander and SchorkLander & Schork, 1994). Examples include coronary heart disease, hypertension, rheumatoid arthritis, type I and type II diabetes mellitus, asthma, many cancers and most psychiatric disorders. In terms of their genetic properties and complexity, psychiatric disorders, including schizophrenia and bipolar disorder, are very similar to the non-psychiatric common familial disorders. In fact, perhaps surprisingly, genetic susceptibility to risk is substantially higher for the major psychiatric illnesses than for most of the non-psychiatric diseases (Reference Plomin, Owen and McGuffinPlomin et al, 1994). What makes the study of psychiatric genetics substantially more difficult than investigation of the complex non-psychiatric diseases is the lack of biologically valid measures for phenotype definition.
RARE VARIANTS OF LARGE EFFECT AND COMMON VARIANTS OF SMALL EFFECT
Theory
McClellan et al consider two distinct genetic models that can explain the transmission of common familial disorders that show non-Mendelian inheritance. They conclude that most cases of schizophrenia are likely to be explained by genetic variants of large effect that, although individually rare, in their totality account for the majority of cases in the population. According to this view, only one rare variant of large effect is involved in each family, but different variants, which may be in the same or in other genes, operate in other families. This is sometimes called the ‘common disease–rare variant' model. It can be contrasted with the ‘common disease–common variant' model which forms the rationale for the large-scale genetic association studies that are ongoing in many centres around the world. In the latter model, a common disease, such as schizophrenia, results from the coaction of multiple (ranging in principle from a few to many thousand) common variants (‘polymorphisms’), each of which has a small effect on illness susceptibility. When an individual inherits several, or many, susceptibility variants together, they have a sizable influence on disease risk. This is essentially the traditional ‘multifactorial’ model that assumes the action of multiple genes and environmental risk factors (Reference Falconer and McKayFalconer & MacKay, 1995).
Schizophrenia
McClellan et al argue strongly against the common disease–common variant model but argue in favour of rare variants of large effect. Since we still do not know the true genetic architecture of schizophrenia, challenges to widely held assumptions and discussions of the possible impact on gene discovery are welcome. However, a considerable body of genetic epidemiological and molecular data relating to schizophrenia as well as population genetic findings do allow some inferences to be drawn and constrain the nature of plausible models. We agree with McClellan et al that rare mutations are likely to be important in some cases of schizophrenia; there are indeed examples in which schizophrenia is related to chromosomal abnormalities. However, it is our contention that key genetic epidemiological and molecular genetic observations are inconsistent with the hypothesis that rare variants of large effect can explain the majority of cases of schizophrenia. Further, the dismissal by McClellan and colleagues of the importance of variants of modest or small effect is not well founded. Important pieces of evidence that contradict their assertions are given below.
Families with clear Mendelian inheritance patterns are rare
Under the model of rare variants of major effect, even allowing for a high proportion of new mutations, it would be expected that there would be many families with clear-cut single gene inheritance. However, as experienced clinical psychiatrists will know, such families are rare.
Single genes of major effect have not been found
Over the past 20 years hundreds of diseases with Mendelian inheritance have been subjected to genetic analysis (‘positional cloning’) which has allowed detection of mutations of major effect using only a few families or just one large pedigree (Reference CollinsCollins, 1992; Reference Botstein and RischBotstein & Risch, 2003), some of which are cited by McClellan et al. Although rare, extended pedigrees multiply affected by psychosis do exist and have been studied genetically. If single genes of major effect explained illness in such pedigrees, the genetic methods used should have identified them, or at least unambiguously defined a chromosomal location, as has been done successfully for the many disorders with Mendelian inheritance. However, when extended pedigrees with multiple cases of illness that are consistent with simple Mendelian inheritance patterns have been subjected to intensive molecular genetic study, not only have mutations of major effect not been identified, the evidence for linkage is much weaker than one would expect if the families were segregating a single cause of the disorder. Rather, findings to date are consistent with multiple variants of modest effect (see Reference Chumakov, Blumenfeld and GuerassimenkoChumakov et al, 2002; Reference Stefansson, Sigurdsson and SteinthorsdottirStefansson et al, 2002; Reference Straub, Jiang and MacleanStraub et al, 2002).
Mathematical modelling of familial risk is inconsistent with single genes of large effect
McClellan et al cite Risch's studies modelling the way risk of illness changes as a function of genetic relatedness to a sufferer (Reference RischRisch, 1990). For both schizophrenia and bipolar disorder there is a very rapid, non-linear decrease of risk when moving from a genetically identical individual (i.e. monozygotic co-twin where the risk is 50–60%), to an individual who shares half the genes (e.g. sibling, parent, dizygotic co-twin where risk is around 10%). Contrary to the assertion by McClellan et al, mathematical modelling demonstrates that this pattern cannot be explained by a collection of genes of large effect that act on their own, even if a sizable proportion are de novo mutations. For illnesses where one mutation is a sufficient cause of illness in each family (whether or not there is a different mutation or gene involved in different families) there is a more gradual (linear) decrease of risk (Reference McGue and GottesmanMcGue & Gottesman, 1989). In contrast, the rapid, non-linear decrease of risk is compatible with multiple interacting risk factors, albeit of unknown frequency, that individually have modest effects (Reference RischRisch, 1990; Reference Craddock, Khodel and Van EerdeweghCraddock et al, 1995).
Molecular genetic findings are consistent with multiple risk alleles of modest effect
Several genes have been implicated repeatedly as conferring risk for schizophrenia or bipolar disorder. These include dysbindin (DTNBP1)) (Reference Straub, Jiang and MacleanStraub et al, 2002; Reference Williams, O'Donovan and OwenWilliams et al, 2005), neuregulin 1 (NRG1; Reference Stefansson, Sigurdsson and SteinthorsdottirStefansson et al, 2002; Reference Tosato, Dazzan and CollierTosato et al, 2005; Reference Munafo, Thiselton and ClarkMunafo et al, 2006) and d-amino acid oxidase activator (DAOA, G72/G30; Reference Chumakov, Blumenfeld and GuerassimenkoChumakov et al, 2002; Reference Detera-Wadleigh and McMahonDetera-Wadleigh & McMahon, 2006). The patterns of effect sizes and allele frequencies are consistent with the common disease–common variant model and with the positive findings that have been emerging in studies of non-psychiatric complex genetic diseases (Reference ToddTodd, 2006). Estimated effect sizes are all modest, with estimated relative risks (or odds ratios) typically below 2.0. In contrast, no rare alleles of large effect have yet been unequivocally identified, although a few rare chromosomal aberrations have been shown to dramatically increase risk (Reference Craddock, O'Donovan and OwenCraddock et al, 2005).
GENETIC COMPLEXITY OF SCHIZOPHRENIA
As is often the case with dichotomous decisions, choosing between either rare variants of large effect or common variants of small effect is almost certainly oversimplistic. Instead, it is probably more reasonable to assume that the spectrum of mutations for common disease is similar to that of all variants in the human genome. This leads to the expectation of a spectrum of risk variants of varying effect sizes, including both common and rare alleles (Reference Wang, Barratt and ClaytonWang et al, 2005). Note that the spectrum of likely risk alleles also includes rare variants of small or modest effect size, the existence of which might well prove to be a far greater obstacle to gene discovery than rare alleles of large effect size.
It is important to acknowledge that, in addition to the models already discussed, several molecular genetic mechanisms are known that result in complex, non-Mendleian patterns of inheritance for a disorder or trait. Examples include: dynamic mutations (e.g. the expanding trinucleotide repeats that underlie fragile X disorder); genomic imprinting (e.g. Prader–Willi syndrome); and mitochondrial inheritance (e.g. some optic atrophies) as well as other mechanisms involving deletion, insertion or variable repetition of stretches of DNA. Such mechanisms might contribute to the genetic complexity of psychiatric illness and need to be considered in the search for genetic factors that influence susceptibility to schizophrenia (see Reference Margolis, McInnis and RosenblattMargolis et al, 1999; Reference MalaspinaMalaspina, 2001; Reference Singh, Murphy and O'ReillySingh et al, 2002; Reference Ben-Shachar and LaifenfeldBen-Shachar & Laifenfeld, 2004).
PHENOTYPIC COMPLEXITY IN PSYCHIATRIC ILLNESS
For most complex genetic diseases, although pathophysiology is incompletely understood, there are biological measures that can be used to define the phenotype (e.g. blood glucose in diabetes, blood pressure in hypertension or biopsy and histology for cancers). These measures are typically reliable and, importantly, have biological validity. The fundamental importance of phenotype definition and measurement for the success of gene identification in human genetics has long been appreciated (Reference Lander and SchorkLander & Schork, 1994). It is surprising, therefore, that the vast majority of psychiatric genetic studies continue to rely on DSM–IV (or ICD–10) diagnostic categories as if they were proven, valid disease entities. Many researchers have assumed that the effects of heterogeneity would be overcome as the technical advances of molecular genetics allowed increasingly large and powerful studies. However, this might not occur if, as well might be the case, researchers tend to adopt less restrictive, or more ‘pragmatic’, inclusion criteria to facilitate the assembly of larger samples. Genetic research might benefit more from the use of smaller, more homogeneous than larger, more heterogeneous samples (Reference Craddock, Owen and O'DonovanCraddock et al, 2006). This principle is clearly demonstrated by the example of the d-amino acid oxidase activator gene (DAOA) which has been implicated in some, but not all, studies of both schizophrenia and bipolar disorder (Reference Chumakov, Blumenfeld and GuerassimenkoChumakov et al, 2002; Reference Hattori, Liu and BadnerHattori et al, 2003; Detera-Wadeleigh & McMahon, 2006). In a large study we found evidence that the gene confers risk for episodes of pathological mood disturbance irrespective of diagnostic category (Reference Williams, Green and MacgregorWilliams et al, 2006). We found significant association in 706 individuals meeting DSM–IV criteria for bipolar disorder. Among those meeting criteria for schizophrenia there was significant association in 112 who had also experienced major mood episodes but not in 597 without major mood episodes. Further, no association was detectable if the schizophrenia sample was treated as a single homogeneous entity (as is the case in most studies). This suggests that results of studies of DAOA in categorically defined ‘schizophrenia’ samples are dependent upon the proportion of people in the sample that have experienced mood disorder – information that is not usually provided or considered by researchers.
A further striking example is provided by the gene Disrupted in Schizophrenia 1 (DISC1). This was identified by studies of an extended Scottish pedigree in which a spectrum of psychiatric illness, including mood and psychotic diagnoses, co-segregated with a chromosomal translocation (Reference St Clair, Blackwood and MuirSt Clair et al, 1990; Reference Millar, Wilson-Annan and AndersonMillar et al, 2000; Reference Muir, Pickard and BlackwoodMuir et al, 2006). Although the name given to the gene by the research team explicitly refers to schizophrenia, major disorder is actually more strongly linked to the translocation (Reference Blackwood, Fordyce and WalkerBlackwood et al, 2001). We have provided independent evidence from a linkage study of families with schizoaffective disorder that variation at the DISC1 locus influences susceptibility to psychopathology involving disturbances in both mood and psychotic domains (Reference Hamshere, Bennett and WilliamsHamshere et al, 2005).
All the molecular evidence suggests that genetic susceptibility does not respect current operational diagnostic boundaries (Reference Craddock and OwenCraddock & Owen, 2005). This will not surprise psychiatrists. ‘Schizophrenia’ is so broad that it is possible for one sample to be composed of individuals with chronic disability involving cognitive impairment, marked negative features and minimal affective or positive psychotic symptoms whereas another sample could include individuals who are able to function relatively well, with an episodic course and marked affective and positive psychotic symptoms. Self-evidently, unless clinical variation is the consequence of chance or of environmental risk factors, illness in each of the above two samples will reflect the operation of at least some susceptibility alleles not held in common at the same frequency in each group. Since the key to unambiguously identifying a risk factor is replication across different samples, we must move beyond diagnostic categories for describing and analysing samples and routinely consider more detailed measures of lifetime psychopathology. There are substantial theoretical benefits of using endophenotypes (intermediate phenotypes) such as neuroimaging or tests of cognitive function to define more homogeneous groups or to access more directly abnormalities that mediate the effects of genes on psychopathology (Reference JablenskyJablensky, 2006; Reference Braff, Freedman and SchorkBraff et al, 2007). However, these approaches are not without difficulties (Reference Owen, Craddock and O'DonovanOwen et al, 2005) and most samples collected to date for genetic studies have clinical data rather than these extended measures. It is therefore relatively simple and inexpensive to make more effective use of the clinical data that can be used to characterise individuals.
ISSUES THAT AFFECT PHENOTYPIC COMPLEXITY
Much attention has been devoted to genetic issues that can affect comparisons between studies. These include genetic differences between different geographical or ethnic populations (so-called population stratification or structure; see Reference Cardon and PalmerCardon & Palmer, 2003), methods of ascertainment (see Reference McCarthy, Kruglyak and LanderMcCarthy et al, 1998), approaches to dealing with unknown genetic models (see Reference RischRisch, 2000), and genotyping error (see Reference Moskvina, Craddock and HolmansMoskvina et al, 2006). In contrast, similar issues contributing to phenotypic heterogeneity have been less widely considered, although they can cause substantial clinical variability between samples (Table 1). These include:
Table 1 Factors affecting issues of genetic and phenotypic complexity in psychiatric genetic studies
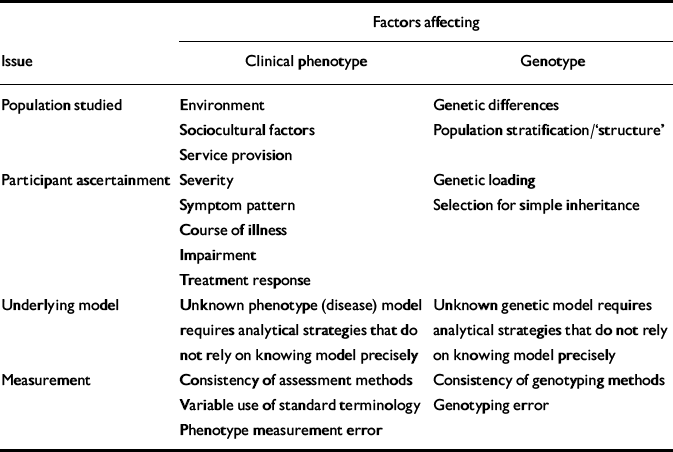
| Factors affecting | |||
|---|---|---|---|
| Issue | Clinical phenotype | Genotype | |
| Population studied | Environment | Genetic differences | |
| Sociocultural factors | Population stratification/‘structure' | ||
| Service provision | |||
| Participant ascertainment | Severity | Genetic loading | |
| Symptom pattern | Selection for simple inheritance | ||
| Course of illness | |||
| Impairment | |||
| Treatment response | |||
| Underlying model | Unknown phenotype (disease) model requires analytical strategies that do not rely on knowing model precisely | Unknown genetic model requires analytical strategies that do not rely on knowing model precisely | |
| Measurement | Consistency of assessment methods | Consistency of genotyping methods | |
| Variable use of standard terminology | Genotyping error | ||
| Phenotype measurement error | |||
-
(a) Geographical origin. In addition to the likelihood that genetic contribution to illness varies between populations, there will be differences resulting from varying environmental exposures, sociocultural factors, service provision, etc.
-
(b) Ascertainment method. The spectrum of clinical features (symptoms, severity, functioning, illness course, etc.) of individuals recruited depends upon the mode of ascertainment. For example, inpatients at a tertiary referral centre differ from out-patients in secondary care.
-
(c) Unknown phenotypic model. Reliance on DSM–IV or ICD–10 categories obscures enormous clinical variability within categories. Perhaps of even greater concern, similarities across different categories of disorder are hidden.
-
(d) Measurement issues. Standardised methodologies for lifetime assessment of psychopathology are available. The same attention that is routinely given to technical issues of laboratory measurement must be given to correct, reliable and consistent use of phenotype measurement.
To maximise the potential of molecular genetic studies we need to pay much more attention to these phenotypic methodological issues than has recently been the norm.
CONCLUSION
There is a need to use a range of genetic approaches that are appropriate to identifying a spectrum of risk variants from the common through to the rare. McClellan et al (Reference McClellan, Susser and King2007) argue for approaches targeting risk alleles of large individual effect but low population effect size. Although data from genetic epidemiology and molecular genetics support the existence of some rare chromosomal abnormalities of large effect size, the evidence suggests rare variants of large effect size do not account for the majority of cases of schizophrenia. However, one unanswered possibility is that most genetic risk results from rare alleles of moderate effect size. Since for the next few years common alleles of modest effect size are likely to be more tractable than are rare alleles, it seems appropriate that the focus in the immediate future will be on large samples and molecular genetic methods powered to detect the common alleles of modest effect size. Such approaches have only become available in the past 1–2 years; it is far too early to judge whether they have been successful or not; indeed, at the time of writing, no whole genome-based surveys for common alleles of moderate effect size have been published. However, we predict that the interpretation of the data from such studies will be impeded by clinical variability across samples.
Replication of novel findings is essential. However, if as we suspect genetic variants that influence risk for psychiatric disorder influence aspects of the phenotype across DSM–IV/ICD–10 categories, and also influence only some aspects of the phenotype within these diagnostic categories, replication in psychiatric genetics will require close attention to both clinical psychiatric methodology and genetic methodology. We will need to become more sophisticated in our phenotypic thinking and move beyond studies and analyses based mainly on the traditional descriptive diagnostic categories (see Reference Craddock and OwenCraddock & Owen, 2005; Reference MarnerosMarneros, 2006; Reference AngstAngst, 2007). We need to ensure that clinical psychiatry is placed very firmly at the heart of psychiatric genetics.


eLetters
No eLetters have been published for this article.