


Schizophrenia is associated with increased use of cannabis and tobacco compared with the general population, although reasons for these associations have not been clearly elucidated. There is some evidence that people with schizophrenia may use tobacco to alleviate neurophysiological deficits associated with this disorder (Reference Adler, Hoffer and WiserAdler et al, 1993; Reference Olincy, Ross and YoungOlincy et al, 1998), and that this is mediated through effects at the α7 nicotinic acetylcholine receptor (CHRNA7) (Reference Gray, Rajan and RadcliffeGray et al, 1996; Reference Stevens, Kem and MahnirStevens et al, 1998). An association between schizophrenia and a putative functional variant, –86C/T, within the CHRNA7 gene (CHRNA7) has been reported (Reference Leonard, Gault and HopkinsLeonard et al, 2002) and warrants further exploration.
The main psychoactive compound within cannabis is delta-9-tetrahydrocannabinol (Δ9-THC), which acts through the CNR1 cannabinoid receptor. An increased incidence of psychotic disorders in people using cannabis has been observed (Reference Arseneault, Cannon and PoultonArseneault et al, 2002; Reference Zammit, Allebeck and AndreassonZammit et al, 2002) and a putative interaction between cannabis use and variation within the catechol-O-methyltransferase (COMT) gene on risk of psychosis has also been reported (Reference Caspi, Moffitt and CannonCaspi et al, 2005). Findings from relatively small studies examining association between CNR1 genetic variation – most commonly at the single nucleotide polymorphism (SNP) rs1049353 – and schizophrenia have been inconsistent, and it was considered worth while to examine this in a substantially larger sample than has been studied thus far.
The main aims of our study were to investigate whether variations at –86C/T within CHRNA7 and at rs1049353 within CNR1 were associated with schizophrenia, and whether these relationships differed according to use of tobacco or cannabis. We also investigated whether there was any evidence of an interaction between cannabis use and the Val158Met polymorphism (SNP rs4680) within COMT, as previously reported (Reference Caspi, Moffitt and CannonCaspi et al, 2005), as well as with SNPs rs737865 and rs165599 within this gene. The SNP rs4680 alters enzyme activity of COMT (Reference Chen, Lipska and HalimChen et al, 2004), whereas the GGG haplotype of SNPs rs737865–4680–165599 has been reported to be associated with lower expression of COMT messenger RNA in human brain tissue (Reference Bray, Buckland and WilliamsBray et al, 2003) and with increased risk of schizophrenia (Reference Shifman, Bronstein and SternfeldShifman et al, 2002). Main genotype and haplotype effects of COMT in this sample have been previously reported, with no evidence found for any association with schizophrenia (Reference Williams, Glaser and WilliamsWilliams et al, 2005).
METHOD
Participants
A sample of unrelated individuals with schizophrenia was recruited from out-patient and in-patient clinical settings and from volunteer support organisations within the UK. These individuals were assessed using the Schedule for Assessment of Neuropsychiatric Disorders semi-structured interview (SCAN; Reference Wing, Babor and BrughaWing et al, 1990) together with case-note review wherever possible. The Operational Criteria Checklist (OPCRIT; Reference McGuffin, Farmer and HarveyMcGuffin et al, 1991) and Global Assessment Scale (GAS; Reference Endicott, Spitzer and FleissEndicott et al, 1976) were also completed. High levels of reliability (κ>0.8) were achieved between raters for diagnoses and rating scale items. Controls were unrelated blood donors ascertained from the same regions as the majority of the patients. Given the prevalence of schizophrenia and the fact that people taking regular medication cannot be blood donors in the UK, it was not deemed necessary to screen the control group for schizophrenia to retain statistical power (Reference Owen, Holmans and McGuffinOwen et al, 1997). Ethical approval was granted for this study and informed consent was obtained from all participants.
All study participants were White, with both parents born in the UK or Ireland. All cases of schizophrenia satisfied DSM–IV criteria (American Psychiatric Association, 1994) for consensus lifetime diagnosis of the disorder, made by two independent raters. The following phenotypes, determined a priori, were examined in relation to –86C/T and rs1049353 genotype:
-
(a) age at onset, defined as the age at which psychiatric help for psychotic symptoms was first sought;
-
(b) worst-ever GAS score, ranging from 0 (most severe) to 100 (least severe);
-
(c) poor outcome, defined as either non-continuous or continuous course of illness;
-
(d) standardised, continuous symptom dimensions obtained by factor analysis of OPCRIT psychosis items, corresponding to paranoid delusions and hallucinations; disorganised symptoms; negative symptoms; and first-rank delusions.
Data on tobacco and cannabis use were obtained from interview and case-note records for 657 of the schizophrenia group. Questions regarding the age at which the person first started using cannabis or tobacco were only introduced during the latter part of the sample recruitment, and these data were therefore available for only 22% of cases in which the person reported ever using these substances. Substance use data were not collected initially for the control group, and unfortunately most members of the control group were not asked at the initial interview for permission to contact them again for further information. As a result of this, cannabis use data were available for only 116 controls and tobacco use data for 49 controls.
Genotyping
The CHRNA7 promoter polymorphism –86C/T was genotyped as a restriction-fragment length polymorphism using the restriction enzyme Hph1 (New England Biolabs, Ipswich, Massachusetts, USA). The primers were 5′-agtacctcccgctcacacctcg-3′ and 5′-atgttgagtcccggagctg-3′ as used by Leonard et al (Reference Leonard, Gault and Hopkins2002). The product was amplified using the GC-RICH PCR System (Roche Diagnostics, Basel, Switzerland), and the 272 base pairs fragment was digested with Hph1 resulting in two fragments of 79 bp and 193 bp with the T allele. The products were run out on a 1.5% agarose gel and visualised using ethidium bromide.
The CNR1 polymorphism rs1049353 was genotyped by fluorescence polarisation using an AcycloPrime kit (PerkinElmer, Waltham, Massachusetts, USA) and the output was read on an LJL Biosystems (Sunnyvale, California, USA) plate reader. A 297 bp amplimere was amplified using primers 5′-ttccctcttgtgaaggcact-3′ and 5′tcattgagcatggtaaagtt-3′. The SNP was at position 125. The extension primer used in the fluorescence polarisation assay was 5′-catggttaccttggcaatcttgac-3′. The COMT markers were genotyped using SNaPshot (Applied Biosystems, Foster City, California, USA) using an ABI3100 sequencer. Details of primers and reaction conditions are provided in Appendix 1 at http://www.cardiff.ac.uk/medicine/psychological_medicine/pub_data/comt.htm.
Analysis
The reference participants for the analyses were those with genotypes that were CC homozygous for –86C/T, GG homozygous for rs1049353, AA homozygous for rs737865, AA homozygous for rs165599, and homozygous for the Met allele at Val158Met within COMT. Only 0.4% of our participants were homozygous for the T allele at the –86C/T locus, and they were therefore grouped with the C/T heterozygotes.
Logistic regression was used to examine associations between dichotomous outcomes and genotypes. A dominance genetic model, as described above, was examined for –86C/T, whereas additive models were used for the CNR1 and COMT variants (Reference LewisLewis, 2002). For the study of continuous phenotypic outcomes, linear regression was used. However, for age at onset, where assumptions of normality were not met, data were ln-transformed prior to regression modelling. Statistical interactions on a multiplicative scale between substance use and genotype on risk of schizophrenia were investigated using a likelihood ratio test within the logistic regression models. For Val158Met, however, as no association was observed between this SNP and cannabis use in the Dunedin cohort (Reference Caspi, Moffitt and CannonCaspi et al, 2005), we used a case-only approach to investigate possible gene–environment interactions because this is statistically more powerful (Reference Khoury and FlandersKhoury & Flanders, 1996). The case-only analysis was also used for rs737865 and rs165599 within COMT. Haplotypes for COMT were examined using UNPHASED, version 3.0 (Reference DudbridgeDudbridge, 2003).
This study had greater than 95% power to detect an additive genetic effect with an odds ratio of 1.4 or above at α=0.05 for the CNR1 and COMT variants examined. This study also had greater than 95% power to find an association between –86C/T variation and schizophrenia based on frequencies of CC genotype of 0.91 in the control group and 0.84 in the schizophrenia group, as observed by Leonard et al (Reference Leonard, Gault and Hopkins2002). The interaction odds ratio previously reported for cannabis and Val158Met was 3.5 (Reference Caspi, Moffitt and CannonCaspi et al, 2005), and our case-only approach had more than 90% power to detect an interaction odds ratio of as low as 1.5, at α=0.05.
Sensitivity analysis
Some participants were likely to have started using tobacco or cannabis after the onset of schizophrenia and it is possible that this could obscure and complicate interpretation of results from this study. Examination of the association between schizophrenia and genotypes was therefore repeated with analyses restricted to cases where the onset of substance use was reported to be at least 1 year prior to age at schizophrenia onset.
RESULTS
There were 838 participants with schizophrenia who were genotyped for any of CNR1 (n=797), CHRNA7 (n=750) or COMT (n=575). Data on cannabis and tobacco use were missing for 96 (11.5%) and 107 (12.8%) of these respectively. Of those with substance use data, 276 participants (37.2%) had ever used cannabis, and 531 (72.6%) had ever used tobacco.
CHRNA7
The –86C/T genotypes were in Hardy–Weinberg equilibrium in both the schizophrenia group (χ2=0.01, P=0.76) and the control group (χ2=0.01, P=0.92). As shown in Table 1, there was no evidence for any association between –86C/T geno-type and schizophrenia (CT/TT genotypes OR= 1.07, 95% CI 0.77–1.49; P=0.70). There was little evidence of any difference in the effect of genotype on schizophrenia between those who smoked (schizophrenia group n=473, controls n=24; OR=3.0, 95% CI 0.4–22.9) and those who did not (schizophrenia group n=186, controls n=25; OR=1.7, 95% CI 0.4–7.7; interaction likelihood ratio test χ2=0.21, d.f.=1, P=0.65). As tobacco use data were available only for a small proportion of the control group, a more powerful case-only analysis was also used, and this also failed to provide any evidence for interaction (n=659; odds ratio for tobacco use by CHRNA7 genotype 0.89, 95% CI 0.53–1.48).
Table 1 Association between CHRNA7 (–86C/T) and CNR1 (rs1049353) genotypes and schizophrenia
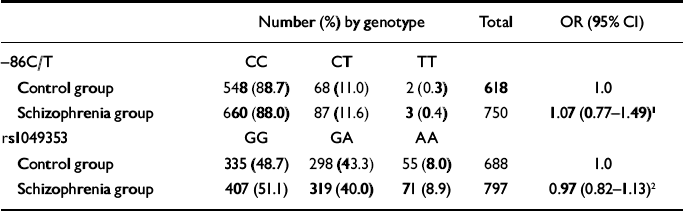
| Number (%) by genotype | Total | OR (95% CI) | |||
|---|---|---|---|---|---|
| -86C/T | CC | CT | TT | ||
| Control group | 548 (88.7) | 68 (11.0) | 2 (0.3) | 618 | 1.0 |
| Schizophrenia group | 660 (88.0) | 87 (11.6) | 3 (0.4) | 750 | 1.07 (0.77-1.49)1 |
| rs1049353 | GG | GA | AA | ||
| Control group | 335 (48.7) | 298 (43.3) | 55 (8.0) | 688 | 1.0 |
| Schizophrenia group | 407 (51.1) | 319 (40.0) | 71 (8.9) | 797 | 0.97 (0.82-1.13)2 |
There were 123 in the schizophrenia group with data relating to age of first using tobacco, and 104 (85%) of these claimed to have started using tobacco prior to the onset of schizophrenia. In the sensitivity analysis there was similarly little evidence of any difference in the effect of genotype on schizophrenia between non-smokers and those smoking prior to illness onset (n=110; OR=2.7, 95% CI 0.3–22.3; interaction χ2=0.1, d.f.=1, P=0.73).
Another way of presenting these data is to examine the relationship between tobacco use and schizophrenia stratified by –86C/T genotype. Tobacco use was strongly associated with schizophrenia in the whole sample (OR=4.4. 95% CI 3.3–6.0; P<0.001), with no evidence of any interaction when stratified by genotype (CC genotype OR=2.6, 95% CI 1.4–4.7; CT/TT genotypes OR=4.6, 95% CI 0.4–53.0; interaction likelihood ratio test as above, P=0.65). Tobacco use was not associated with –86C/T genotype (OR=0.9, 95% CI 0.5–1.5).
Results for associations between –86C/T genotype and various phenotypes within schizophrenia are presented in Table 2. There was weak evidence (P=0.07) that participants with the CT/TT genotypes had a younger age of onset, by approximately 2 years on average, than those homozygous for the C allele.
Table 2 Effect estimates for phenotype characteristics according to CHRNA7 (–86C/T) and CNR1 (rs1049353) genotypes in participants with schizophrenia
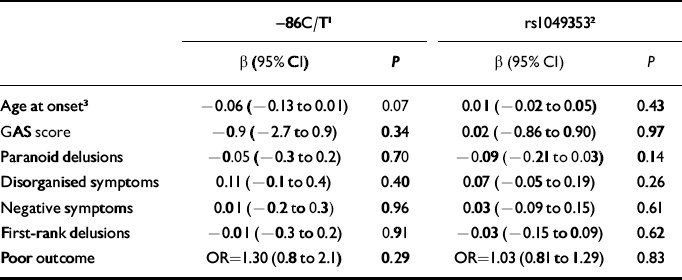
| -86C/T1 | rs10493532 | |||
|---|---|---|---|---|
| β (95% CI) | P | β (95% CI) | P | |
| Age at onset3 | -0.06 (-0.13 to 0.01) | 0.07 | 0.01 (-0.02 to 0.05) | 0.43 |
| GAS score | -0.9 (-2.7 to 0.9) | 0.34 | 0.02 (-0.86 to 0.90) | 0.97 |
| Paranoid delusions | -0.05 (-0.3 to 0.2) | 0.70 | -0.09 (-0.21 to 0.03) | 0.14 |
| Disorganised symptoms | 0.11 (-0.1 to 0.4) | 0.40 | 0.07 (-0.05 to 0.19) | 0.26 |
| Negative symptoms | 0.01 (-0.2 to 0.3) | 0.96 | 0.03 (-0.09 to 0.15) | 0.61 |
| First-rank delusions | -0.01 (-0.3 to 0.2) | 0.91 | -0.03 (-0.15 to 0.09) | 0.62 |
| Poor outcome | OR=1.30 (0.8 to 2.1) | 0.29 | OR=1.03 (0.81 to 1.29) | 0.83 |
CNR1
Genotypes at rs1049353 were in Hardy–Weinberg equilibrium in both the schizophrenia group (χ2= 0.56, P=0.44) and controls (χ2=1.0, P=0.36). As shown in Table 1, there was no evidence for any association between rs1049353 genotype and schizophrenia (odds ratio for linear trend of genotypes 0.97, 95% CI 0.82–1.13; P=0.66).
There was little evidence of any difference in the effect of rs1049353 genotype on schizophrenia between those who did not use cannabis (schizophrenia group n=445, controls n=93; OR=1.04, 95% CI 0.73–1.47) and those who did (schizophrenia group n=261, controls n=23; OR=0.92, 95% CI 0.48–1.75; interaction χ2=0.11, d.f.=1, P=0.74). As cannabis use data were again available for only a small proportion of the control group, a case-only analysis was used, and this also failed to provide any evidence for interaction (n=706; odds ratio for cannabis use by CNR1 genotype 0.83, 95% CI 0.65–1.05).
As part of the sensitivity analysis, there were 71 individuals in the schizophrenia group with data relating to age of first using cannabis, and 64 (90%) of these reported first use prior to onset of schizophrenia. As in the main analysis, there was little evidence of any difference in the effect of rs1049353 genotype on schizophrenia between those who did not use cannabis and those who claimed to have used cannabis at least 1 year prior to illness onset (n=614; OR=0.84, 95% CI 0.40–1.78; interaction χ2=0.26, d.f.=1, P=0.61).
Presenting these data in another way, there was a strong association between cannabis use and schizophrenia in this sample (OR=2.6, 95% CI 1.8–3.7; P<0.001), with no evidence of any difference when stratified by rs1049353 genotype (GG genotype OR=2.3, 95% CI 1.2–4.4; GA genotype OR=3.1, 95% CI 1.3–7.2; AA genotype OR=1.1, 95% CI 0.2–4.6; interaction likelihood ratio test as above, P=0.33). There was no evidence for any association between rs1049353 genotype and various phenotypes within schizophrenia (Table 2).
COMT
There was no evidence for any association between Val158Met genotype and cannabis use in our sample of 493 persons with schizophrenia (OR=0.98, 95% CI 0.76–1.27, P=0.89). Results were almost identical when restricting the analysis to participants who first used cannabis at least 1 year prior to their illness onset and who had first used it by age 18 years or earlier (n=338; OR=0.76, 95% CI 0.41–1.40; P=0.38). Similarly, there was no evidence that variation at rs737865 or rs165599 was associated with cannabis use in the case-only analysis, even when restricting the analysis to first use of cannabis at least 1 year prior to illness onset and first use by age 18 years or earlier (rs737865, OR=1.09, 95% CI 0.56–2.00; rs165599, OR=1.09, 95% CI 0.57–2.08). There was no evidence of overall haplotype association with cannabis use (χ2=4.7, d.f.=7, P=0.69) or of specific association with the rs737865–4680–165599 GGG haplotype (χ2=0.001, d.f.=1, P=0.98).
DISCUSSION
There was no evidence for any association between CHRNA7 or CNR1 genotype and schizophrenia in our sample, and also no evidence of any gene–environment interactions between tobacco use and CHRNA7, or cannabis use and CNR1 or COMT genotype.
CHRNA7 and tobacco use
There have been few association studies of polymorphisms within CHRNA7 and schizophrenia to date. Leonard et al (Reference Leonard, Gault and Hopkins2002) screened the core promoter region of the full-length gene and reported an association between schizophrenia and variant –86C/T. Although we found no evidence for an association between the promoter SNP –86C/T and schizophrenia, CT/TT genotypes occurred slightly more frequently in participants with schizophrenia than in controls, in a direction consistent with the findings by Leonard et al (Reference Leonard, Gault and Hopkins2002). However, we observed a much smaller difference in CC frequency of less than 1%, as opposed to the 7% reported in the original study (Reference Leonard, Gault and HopkinsLeonard et al, 2002).
People with schizophrenia commonly display evidence of sensory attention impairments (Reference Adler, Pachtman and FranksAdler et al, 1982; Reference Leonard, Adams and BreeseLeonard et al, 1996), including deficits in pre-pulse inhibition and P50 gating response (Reference Braff and SaccuzzoBraff & Saccuzzo, 1985; Reference Braff, Grillon and GeyerBraff et al, 1992; Reference Waldo, Cawthra and AdlerWaldo et al, 1994). Improvements in such neurophysiological deficits in people with schizophrenia following cigarette smoking have been reported (Reference Adler, Hoffer and WiserAdler et al, 1993; Reference Olincy, Ross and YoungOlincy et al, 1998), with similar improvements observed following nicotine administration in animal models (Reference Bickford and WearBickford & Wear, 1995; Stevens et al, Reference Stevens, Freedman and Collins1996, Reference Stevens, Kem and Mahnir1998). Specific agonists of the α7 receptor (CHRNA7) normalise sensory gating deficits in animal models (Reference Stevens, Kem and MahnirStevens et al, 1998), whereas evidence for genetic linkage to the P50 deficit and, to a lesser extent, to schizophrenia, has been reported for chromosome band 15q14, an area that contains CHRNA7 (Reference Coon, Plaetke and HolikCoon et al, 1993; Reference Freedman, Coon and Myles-WorsleyFreedman et al, 1997; Reference Leonard, Gault and MooreLeonard et al, 1998).
Despite this support, from a variety of sources, that CHRNA7 is a good candidate gene for schizophrenia, there is weak evidence at present that variation within this gene is associated with the disorder (Reference Riley, Makoff and Mogudi-CarterRiley et al, 2000; Reference Xu, Pato and TorreXu et al, 2001; Reference Leonard, Gault and HopkinsLeonard et al, 2002; Reference Gault, Hopkins and BergerGault et al, 2003; Reference Li, Liao and ChenLi et al, 2004; Reference Fan, Ma and LiFan et al, 2006). However, given the findings from experimental studies of the effect of nicotine on neurophysiological deficits in both animal models and humans, as described earlier, it may be that any association between CHRNA7 and schizophrenia is mediated by impairments in sensory gating or other related physiological responses. In the study by Leonard et al (Reference Leonard, Gault and Hopkins2002), presence of the T allele at –86C/T was also associated with reduced inhibition of the P50 response in the control group, and although two other studies did not replicate this finding (Reference Gault, Hopkins and BergerGault et al, 2003; Reference Houy, Raux and ThibautHouy et al, 2004), one reported an association between P50 sensory gating response and another promoter variant, –194G/C (Reference Houy, Raux and ThibautHouy et al, 2004). There is a clear need for research into CHRNA7 variation in relation to neurophysiological deficits in well-designed and adequately powered studies to address this further.
CNR1, COMT and cannabis
We found no evidence of association between the CNR1 locus rs1049353 and schizophrenia, consistent with the overall findings previously reported for this variant from two much smaller studies (Reference Leroy, Griffon and BourdelLeroy et al, 2001; Reference Ujike, Takaki and NakataUjike et al, 2002), although one of these reported an association in a subgroup analysis (Reference Leroy, Griffon and BourdelLeroy et al, 2001). Two studies have reported associations between schizophrenia and variation within an (AAT) n microsatellite approximately 20 kb upstream of the translational start site of CNR1 (Reference Ujike, Takaki and NakataUjike et al, 2002; Reference Martinez-Gras, Hoenicka and PonceMartinez-Gras et al, 2006). However, different alleles were associated with increased risk in these two studies, and the association in one of the studies was again observed only for a subgroup of participants, this time with hebephrenic schizophrenia.
The CNR1 gene is located on 6q14–15, a region of replicated linkage for schizophrenia (Reference Lewis, Levinson and WiseLewis et al, 2003). There are four SNPs within CNR1 on HapMap that have a heterozygosity in European populations greater than 0.1; three of these are in the 3′ untranslated region whereas rs1049353 is a synonymous SNP found within exon 1. The relatively small size of CNR1, the limited variation within the gene and its linkage disequilibrium structure mean it is unlikely that a substantial effect on schizophrenia risk is conferred by variation within this gene, given our findings and the lack of other consistent associations reported to date.
We also failed to find any supporting evidence for a differential effect of cannabis use on psychosis risk according to variation at Val158Met within COMT. In the Dunedin study evidence for an interaction was observed only for people first using cannabis by age 18 years, but not for those using it after this age (Reference Caspi, Moffitt and CannonCaspi et al, 2005). One explanation proposed for this was that there may exist a sensitive or even critical period of risk when the influence of cannabis exposure is moderated by COMT genotype. In our study we failed to find evidence for an interaction between cannabis use and COMT genotype even when restricting the analysis to participants who claimed to have first used cannabis by the same cutoff period of age 18 years, despite more than adequate statistical power to replicate the original findings. Furthermore, in contrast to the findings by Caspi et al (Reference Caspi, Moffitt and Cannon2005), cannabis use by age 18 years was actually less common in participants with schizophrenia homozygous for the Val allele compared with those heterozygous for this allele or homozygous for Met (5.3%, 6.4% and 8.7% respectively), although this was not significantly different.
Limitations to the interpretation of our results
This study was adequately powered to examine main effects on risk of schizophrenia, suggesting it is unlikely that variations in CNR1 or CHRNA7 are important risk factors for schizophrenia. Furthermore, this study was adequately powered for studies of interactions using a case-only design, but this approach is dependent on the assumption of no genotype–exposure association in the population. For COMT this assumption is likely to be a reasonable one, given that no association with cannabis use was observed in the Dunedin cohort (Reference Caspi, Moffitt and CannonCaspi et al, 2005). However, this assumption may be less likely to hold true for CNR1 or CHRNA7, given that cannabis and nicotine act through receptors coded for by these genes, and also given the sporadic reports of associations between cannabis and tobacco dependence and CNR1/CHRNA7 genotypes (Reference Greenbaum, Kanyas and KarniGreenbaum et al, 2006; Reference Hopfer, Young and PurcellHopfer et al, 2006). For that reason we also conducted studies of interactions between CNR1 and cannabis as well as between CHRNA7 and tobacco using a more traditional case–control approach, although statistical power to exclude anything other than large interaction effects for these two genes using this latter approach was limited.
Although we genotyped three SNPs in COMT that together form a haplotype reported to be significantly associated with schizophrenia (Reference Shifman, Bronstein and SternfeldShifman et al, 2002), we only genotyped one SNP in each of CNR1 and CHRNA7. It is not possible therefore to rule out causal effects of variants within these genes that are not in strong linkage disequilibrium with the SNPs we tested. However, a strong effect of CNR1 on risk of schizophrenia seems unlikely, given the linkage disequilibrium structure within this gene. Our confidence in ruling out such an effect for CHRNA7 is lower, although we did not feel that the evidence we obtained was strong enough to warrant further genotyping of CHRNA7 SNPs, especially given the problems resulting from the partial duplication of this gene, which makes such studies inherently more difficult.
A final limitation of our study is that, unlike the longitudinal data collection in the Dunedin cohort, our case–control design relied on people recalling age of first use of cannabis and relating this in time to the date of their first contact with psychiatric services. Such data seem inherently more likely to be misclassified than prospectively collected data. It is unclear to what extent any such misclassification might have resulted in an underestimate of the association between cannabis use and genotype in our case-only analysis, and therefore obscured any true interaction effect. It would, however, presumably require a substantial amount of misclassification to obscure an interaction effect as strong as that reported by Caspi and colleagues, whereby cannabis use was associated with a 10-fold increase in risk of psychotic disorder in those homozygous for valine but had no effect in those homozygous for methionine (Reference Caspi, Moffitt and CannonCaspi et al, 2005). This finding of an interaction effect in the Dunedin cohort was observed only in a subgroup of participants – those using cannabis by age 18 years. Similarly, supportive evidence of a putative interaction between cannabis use and COMT on psychotic symptoms, following administration of cannabis in an experimental setting was again observed only in a subgroup of participants with schizophrenia, this time those with evidence of pre-existing psychotic traits (Reference Henquet, Rosa and KrabbendamHenquet et al, 2006). Although such findings are biologically plausible and seem intuitively appealing, substantially more evidence from replication of these findings is required. Our study, although providing adequate power to observe even a relatively small association between cannabis use and COMT genotype in participants with schizophrenia, may not be the ideal design to examine such a relationship, and other longitudinal studies may be able to investigate this with greater confidence in the future.
In summary, we failed to find any evidence that variation at the CHRNA7 or CRN1 locus was associated with schizophrenia, or that the effect of variation at these loci was modified by use of tobacco or cannabis respectively. Cannabis use was not associated with presence of the valine allele at Val158Met within COMT in our sample, therefore our findings do not not support a previous report of a putative gene–environment interaction between COMT genotype and cannabis use on risk of schizophrenia.
Acknowledgements
S.Z. is funded through a Clinician Scientist Award from the National Assembly for Wales.



eLetters
No eLetters have been published for this article.