


Several lines of evidence suggest that abnormal growth during development is associated with an increased risk of adult schizophrenia. A small high-risk sample showed that slower growth in childhood is associated with early-onset schizophrenia (Reference FishFish, 1959). However, results are inconsistent in other studies and most are limited by reliance on isolated (rather than serial) measurements of height. Large cohort studies extrapolating growth patterns from measures of birth weight, birth length and adult weight and height suggest that atypical growth is associated with adult psychosis (Gunnell et al, Reference Gunnell, Rasmussen and Fouskakis2003, Reference Gunnell, Harrison and Whitley2005a ). Such associations suggest that factors responsible for abnormal growth might influence the pathogenesis of schizophrenia. We compared patterns of growth using serial measurements between individuals with schizophrenia and healthy participants from a large and well-characterised birth cohort (Reference Susser, Schaefer and BrownSusser et al, 2000). We predicted that adults who went on to develop schizophrenia would exhibit a slower rate of growth during early life and later childhood, and that the effect would be modified by gender.
METHOD
The cohort comprised offspring of women participating in the Kaiser Foundation Health Plan (KFHP) who received obstetric care and were delivered in Alameda County, California, between 1959 and 1967, and who agreed to participate in the Child Health and Development Study (CHDS) (n=19 044). The KFHP was one of the first and largest health insurance plans in the USA, and nearly all pregnant women who were members participated in the CHDS. A comparison of these participants' demographic data with US census data demonstrated that although both income extremes were underrepresented, members were demographically similar with respect to ethnicity, occupation and educational attainment (Reference KriegerKrieger, 1992).
The study reported here is restricted to a cohort referred to as the Prenatal Determinants of Schizophrenia (PDS) study cohort, which is a subsample of the offspring of those participating in the CHDS. The design for the PDS study cohort has been previously described in full (Reference Susser, Schaefer and BrownSusser et al, 2000); thus, only a summary is provided here. The PDS study cohort was designed to follow up and assess participants for the presence of schizophrenia-spectrum disorders in order to evaluate developmental determinants of schizophrenia. It included offspring of mothers from the CHDS cohort who were members of KFHP from 1 January 1981 to 31 December 1997 (n=12 094). These dates correspond to the period of case ascertainment, which commenced when KFHP began using computerised records, making it feasible to identify all members who had accessed mental health services. Participants in the PDS cohort are comparable with the CHDS sample, with two exceptions: mothers of African American ethnicity were more likely to be included in the PDS cohort (CHDS 16%, PDS 28%) and offspring of low-income unmarried mothers were somewhat less likely to be included (CHDS 26, PDS 20%).
Height measurements
Height in inches to the nearest sixteenth of an inch (1.6 mm) was measured and recorded during regular paediatric visits from birth to age 13 years. These measurements were systematically abstracted from medical records by trained CHDS abstractors. The present analyses were restricted to data from birth to age 9 years, since reliable data on the pubertal stage of development were not available. The average number of height measurements between birth and age 2½ years was 9 for both the schizophrenia-spectrum disorder (SSD) group and the non-SSD group ranging from 3 to 15 measurements for the former and 1 to 31 for the latter. Between ages 2½ years and 9 years the average number of height measurements was 6 for both groups, ranging from 1 to 19 measurements for the SSD group and 1 to 41 for the non-SSD group. To identify outliers, height was standardised by computing gender- and age-adjusted means using the least mean squares (LMS) method (Reference ColeCole, 1990). This method is advantageous for standardising height because it uses a series of calculations to reduce asymmetry in skewed data. Height measurements that were greater than 4 standard deviations above or below the mean were considered to be outliers and were excluded from the analyses; 1.0% of all measurements were excluded.
Assessment of potential confounders
Potential confounding factors were determined a priori based on characteristics that have been shown to be associated with both height and schizophrenia. These included gender, maternal race and education, standardised maternal height, pre-pregnancy body mass index (BMI), gestational age at birth and birth weight. (It is unnecessary to control for birth length in the adjusted analysis because it is used to estimate growth patterns.) Demographic measures were assessed through maternal interview, which was completed during the first prenatal visit.
Maternal race was categorised as White, Black or other. Maternal education was rated on a seven-point scale reflecting the highest level of education achieved. Because not all levels of education were represented among the SSD sample, the categories were collapsed as follows: less than high-school diploma, high-school graduate with or without trade school, high-school graduate plus 1–3 years of college, and college graduate.
Maternal height was measured during the maternal interview. Because paternal height was based on maternal report and we had insufficient data on paternal height, standardised maternal height was used as a proxy for the child's genetic growth potential. Standardisation of maternal height was accomplished using the LMS method. Because adult height is not typically achieved until 20 years of age, z-score transformations of height for mothers aged 15–19 years were calculated separately at 1-year intervals. Mothers aged 20 years or over were assumed to have attained their adult height and were standardised as one group irrespective of age. Maternal pre-pregnancy BMI was calculated using self-reported weight prior to pregnancy and was classified based on categories used in a previous study assessing the association between maternal BMI and schizophrenia-spectrum disorders in this cohort: low, ⩽19.9 kg/m2; average, 20.0–26.9 kg/m2; greater than average, 27.0–29.9 kg/m2; high, ⩾30.0 kg/m2) (Reference Schaefer, Brown and WyattSchaefer et al, 2000). Maternal pre-pregnancy BMI data were missing in a proportion of the sample (8 SSD and 1166 non-SSD). To preserve sample size, the average maternal BMI value was substituted for missing data in the adjusted analyses. Gestational age at birth was calculated as the number of days between the last reported menstrual period and birth.
Ascertainment and diagnosis
We identified potential cases of schizophrenia-spectrum disorder through the KFHP computerised records of in-patient, out-patient and pharmacy registries. With regard to the hospitalisation registry, potential cases were first identified if the individual had received ICD–9 diagnosis codes of 295, 296, 297, 298 or 299 (World Health Organization, 1978) or were not given a specific diagnosis. A review of psychiatric and medical records by a psychiatrist was conducted to determine whether individuals screened positive for evidence of a psychotic disorder. Individuals from the out-patient registry were considered to be screen-positive for SSD if they had diagnosis codes of 295, 297, 298 or 299. For the pharmacy registry, cases screened positive if the individual had received treatment with antipsychotic medication.
We identified 183 participants for further diagnostic assessment. Of those identified, 13 had died. Of the 170 remaining, 146 (86%) were successfully contacted and 107 (58% of those originally identified) completed the Diagnostic Interview for Genetic Studies (DIGS; Reference Nurnberger, Blehar and KaufmannNurnberger et al, 1994) administered by a trained research clinician. For the remaining 76 (42%) individuals who were not interviewed, a diagnosis was made based on review of the medical records by trained clinicians. All individuals provided written informed consent for participation prior to the diagnostic interview. Informed consent was approved by the institutional review boards of the New York State Psychiatric Institute and the Kaiser Permanente Division of Research. Diagnoses were made by consensus of three diagnosticians who independently reviewed all relevant material for each case. In total, 71 cases were identified (43 schizophrenia, 17 schizoaffective disorder, 5 schizotypal disorder, 1 delusional disorder and 5 other schizophrenia-spectrum psychosis). Forty-four individuals completed the DIGS and 27 were diagnosed by chart review.
Analytical data-set
The PDS cohort comprised 12 094 individuals −71 SSD and 12 023 non-SSD. Because our analysis required information obtained through maternal interview during enrolment, people who had not completed the interview were excluded (n= 2412), reducing the sample size to 9682. Since siblings represent non-independent observations, only one sibling per family was included in the analysis and siblings of offspring with schizophrenia-spectrum disorder were excluded (n=1886). Two people in the SSD group were siblings, so one of them was randomly selected and the data excluded from the analysis. The final cohort consisted of 7795 persons. Please see Susser et al (Reference Susser, Schaefer and Brown2000) for a further description of the method for deriving the analytic sample. From this cohort only 15 individuals (all from the non-SSD group) were excluded because they did not have at least one valid height measurement; thus, the analytic sample comprised 7780 persons (SSD n=70; non-SSD n=7710). A summary of the exclusion criteria for the analytical data-set is provided in Fig. 1.
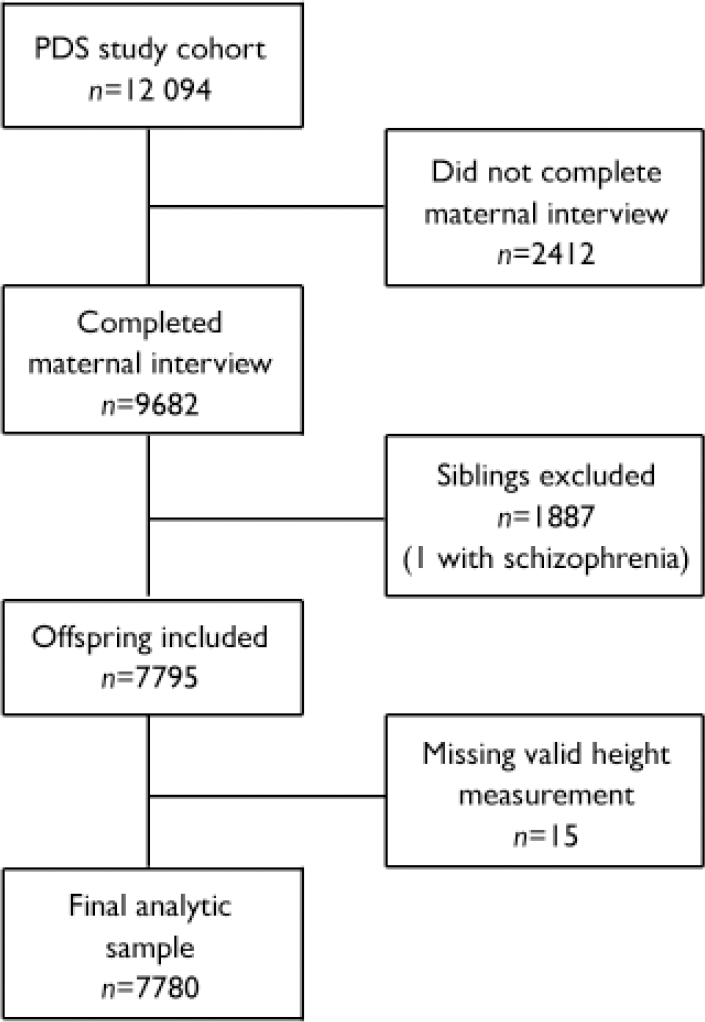
Fig. 1 Study profile (PDS, Prenatal Determinants of Schizophrenia).
Statistical analyses
The relationship between growth and adult schizophrenia was examined using multilevel growth models (Reference McArdle, Aber, Little, Schnabel and BaumertMcArdle & Aber, 2000; Reference Cohen, Cohen and WestCohen et al, 2003; Reference Chen and CohenChen & Cohen, 2006). The PROC MIXED function in the SAS statistical package (Reference Littell, Milliken and StroupLittell et al, 1996) was used to estimate the growth pattern of height. In these multilevel growth models, longitudinal data on individuals are considered the basic ‘random’ data, similar to a cross-sectional study where single individual variables are the basic units of analyses (Reference Cohen, Cohen and WestCohen et al, 2003).
The first step in the analyses was to determine the best model fit for the relationship between growth and age from birth to 2½ years, which is referred to as the basic growth model. We first examined the mean level model, which is the between-participant differences in mean level of height (i.e. height=β0+error). The results suggested that between participants differences were found for height; the estimated variance of the mean height was 2.06 (s.e.=0.28, P < 0.001). To determine the best model for predicting growth, we compared three additional models: linear change in height (height=β1+age+error), quadratic change in height (height=β2+ age+age2+error) and cubic change in height (height=β3+age+age2+age3+error), where random variation was permitted for each age term. To estimate model fit, we calculated the chi-squared value by subtracting the −2 log likelihood estimates from the subsequent models (i.e. mean level v. linear, linear v. quadratic, and quadratic v. cubic). Compared with the mean level model, the addition of the linear term significantly improved model fit (P < 0.001). The addition of the quadratic term also significantly improved model fit compared with the linear model (P < 0.001). Also, the cubic model appeared to significantly improve model fit compared with the quadratic model (P < 0.001). However, the fixed effect of the cubic term was small and the significant improvement mostly due to the random effects. Also, the addition of the cubic term reduced the variance associated with the linear term to zero. Therefore, to facilitate interpretation, and in the interest of comparability and parsimony, we chose to use the quadratic model. Although fractional polynomial models are sometimes used in this type of analysis, a quadratic equation appears to be an accurate estimate of growth and is easier to interpret. In summary, the model comparisons suggested that the quadratic model provided the best model fit for estimating patterns of growth.
Once the basic model for growth was established, we fitted a conditional model to examine the effect of schizophrenia-spectrum disorders on growth. We added SSD (1, case, 0, non-case) as a covariate to examine mean level differences in height. To assess growth differences between the SSD and non-SSD groups, we included an interaction term for age and SSD and an interaction term for (age)2 and SSD in the quadratic model. All adjusted analyses controlled confounding effects of gender, maternal education, race, BMI and height, gestational age at birth and birth weight. Each model included a χ2-test of improvement of fit to the data.
Using the methods described above we also estimated the basic model for growth from age 2½ to 9 years. Separate analyses were performed for early life (birth to age 2½ years) and later childhood (2½ to 9 years) because growth during these periods is regulated by different mechanisms (Reference KarlbergKarlberg, 1987; Reference Reiter, Rosenfeld, Larsen, Kronenberg and MelmedReiter & Rosenfeld, 2003). Additionally, previous research suggests that growth during the first 2 years of life is more variable than growth in later childhood and abnormal growth patterns may be indicative of problems during prenatal development (Reference TannerTanner, 1994; Reference Reiter, Rosenfeld, Larsen, Kronenberg and MelmedReiter & Rosenfeld, 2003). Hence, we predicted that early life (birth to 2½ years) would be associated with the most pronounced growth deficit in individuals who later developed schizophrenia. Previous studies also demonstrated gender differences in growth patterns in healthy samples (Reference KarlbergKarlberg, 1987) and in the risk and correlates of schizophrenia (Reference Goldstein, Seidman and O'BrienGoldstein et al, 2002; Reference Aleman, Kahn and SeltenAleman et al, 2003). Thus, all analyses were performed on the entire cohort as well as being further stratified by gender in order to determine whether differences in growth patterns between individuals in the SSD and non-SSD groups varied by gender.
In addition to assessments using the multilevel growth model, we calculated the linear slope (linear change in height by age) for each participant by using maximum likelihood estimates from the basic growth models for both early life (birth to age 2½ years) and later childhood (2½ to 9 years). A feature of this program is the ability to output the empirical Bayesian estimates of the linear slope of height for each participant over the defined periods (Reference McArdle, Small and BäckmanMcArdle et al, 2005). The linear slopes provide additional information about differences between the SSD and non-SSD groups observed from the multilevel models.
RESULTS
Sample characteristics are summarised in Table 1. Men were overrepresented in the SSD group, whereas gender was more evenly distributed among the cohort sample. The SSD sample included a higher proportion of Black people. The mothers of those in the SSD group had fewer years of education, and their maternal pre-pregnancy BMI was higher. There was no difference in maternal height between the SSD and non-SSD groups and maternal height was normally distributed in both groups. Gestational age at birth was slightly higher in the SSD sample but the groups did not differ with respect to birth length or weight.
Table 1 Sample characteristics (n=7780)

| SSD group (n=70) | Non-SSD group (n=7710) | |
|---|---|---|
| Gender, n (%) | ||
| Male | 46 (66) | 3932 (51) |
| Female | 24 (34) | 3778 (49) |
| Maternal ethnicity, n (%) 1 | ||
| White | 34 (49) | 5018 (65) |
| Black | 31 (45) | 2091 (27) |
| Other | 4 (6) | 592 (8) |
| Maternal education, n (%) 2 | ||
| Less than high school | 15 (25) | 1374 (18) |
| High school, trade school | 25 (41) | 3050 (40) |
| High school plus some college | 12 (20) | 1930 (25) |
| College graduate | 9 (15) | 1341 (17) |
| Maternal BMI, n (%) 3 | ||
| Low (19.9 kg/m2 or less) | 14 (23) | 1529 (23) |
| Average (20.0–26.9 kg/m2) | 37 (60) | 4462 (68) |
| Above average (27.0–29.9 kg/m2) | 5 (8) | 366 (6) |
| High (30.0 kg/m2 or above) | 6 (10) | 187 (3) |
| Gestational age at birth (days): mean (s.d.) 4 | 283 (14) | 280 (16) |
| Maternal height 5 | ||
| Height, cm: mean (s.d.) | 163.36 (6.19) | 162.46 (6.39) |
| z-score: mean (s.d.) | 0.14 (0.96) | –0.01 (0.99) |
| Birth length 6 | ||
| Length, cm: mean (s.d.) | 51.59 (2.77) | 51.52 (2.61) |
| z-score: mean (s.d.) | –0.01 (1.07) | 0.00 (0.99) |
| Birth weight 7 | ||
| Weight, kg: mean (s.d.) | 3.35 (0.54) | 3.33 (0.5) |
| z-score: mean (s.d.) | 0.02 (1.04) | 0.00 (0.98) |
| Height at age 2½ years 8 | ||
| Height, cm: mean (s.d.) | 89.47 (3.34) | 91.20 (3.73) |
| z-score: mean (s.d.) | –0.43 (0.81) | 0.01 (1.00) |
| Height at age 9 years 9 | ||
| Height, cm: mean (s.d.) | 128.43 (3.83) | 132.74 (6.30) |
| z-score: mean (s.d.) | –0.64 (0.53) | –0.01 (0.99) |
| BMI at age 2½ years 10 | ||
| BMI, kg/m2: mean (s.d.) | 16.76 (1.68) | 16.61 (1.41) |
| z-score: mean (s.d.) | 0.05 (1.20) | –0.01 (1.00) |
| BMI at age 9 years 11 | ||
| BMI, kg/m2: mean (s.d.) | 16.83 (1.00) | 17.33 (2.62) |
| z-score: mean (s.d.) | 0.06 (0.38) | –0.00 (1.00) |
BMI, body mass index; SSD, schizophrenia-spectrum disorder
1. Missing/invalid data: SSD group, n=1; non-SSD group, n=9
2. Missing/invalid data: SSD group, n=9; non-SSD group, n=15
3. Missing/invalid data: SSD group, n=8; non-SSD group, n=1166
4. Missing/invalid data: SSD group, n=1; non-SSD group n=78
5. Missing/invalid data: SSD group, n=3; non-SSD group, n=137
6. Missing/invalid data: non-SSD group, n=29
7. Missing/invalid data: non-SSD group, n=26
8. Missing/invalid data: SSD group, n=60; non-SSD group, n=6258
9. Missing/invalid data: SSD group, n=65; non-SSD group, n=7086
10. Missing/invalid data: SSD group, n=59; non-SSD group, n=6263
11. Missing/invalid data: SSD group, n=65; non-SSD group, n=7091
Change in height
The first step in the analyses was to determine the best model fit for estimating growth in our cohort, which is referred to as the basic model. As noted in the statistical analysis section, we determined that the quadratic model, allowing between-participant variance, provided the best model fit for estimating growth (i.e. height=β0+age+age2+error). With respect to the early-life model, the average height at birth for the entire sample was 52.06 cm (Table 2). On average, female babies were 0.82 cm shorter at birth than the males (51.64 cm and 52.46 cm respectively). For the entire sample there was a linear increase with age of 30.27 cm per year (Table 2). Rates of growth declined with age and were best described by a model with a linear and quadratic term. In accordance with normal growth patterns (Reference KarlbergKarlberg, 1987), growth during childhood was slower than during early life and the girls remained shorter than the boys (Table 2).
Table 2 Estimated fixed effects assessing the basic growth model

| Basic growth model | |||||
|---|---|---|---|---|---|
| B | 95% CI | P | |||
| Early life (birth to 2½ years) | |||||
| Entire sample (n=7780) | |||||
| Mean height at birth, cm (intercept) | 52.06 | 52.01 to 52.11 | <0.001 | ||
| Linear change in height, cm/year | 30.27 | 30.12 to 30.37 | <0.001 | ||
| Quadratic change in height, cm/year2 | –6.69 | –6.73 to −6.64 | <0.001 | ||
| Females only (n=3802) | |||||
| Mean height at birth, cm (intercept) | 51.64 | 51.57 to 51.72 | <0.001 | ||
| Linear change in height, cm/year2 | 29.53 | 29.39 to 29.67 | <0.001 | ||
| Quadratic change in height, cm/year2 | –6.37 | –6.44 to −6.30 | <0.001 | ||
| Males only (n=3978) | |||||
| Mean height at birth, cm (intercept) | 52.46 | 52.39 to 52.54 | <0.001 | ||
| Linear change in height, cm/year | 30.95 | 30.80 to −31.10 | <0.001 | ||
| Quadratic change in height, cm/year2 | –6.98 | –7.04 to −6.91 | <0.001 | ||
| Later childhood (2½–9 years) | |||||
| Entire sample (n=7353) | |||||
| Mean height at age 2½ years, cm (intercept) | 90.51 | 90.42 to 90.61 | <0.001 | ||
| Linear change in height, cm/year | 7.97 | 7.92 to 8.02 | <0.001 | ||
| Quadratic change in height, cm/year2 | –0.22 | –0.23 to −0.22 | <0.001 | ||
Association between potential confounders and height
We found the expected relationships between the confounders and variability in height at birth and age 2½ years. Compared with the White participants, Black participants were significantly shorter at birth but taller at age 2½ years. Mothers with a higher level of educational attainment tended to have offspring who were taller at birth and at age 2½ years. Pre-pregnancy maternal BMI was positively correlated with birth length, and greater gestational age at birth was associated with increased birth length and height at age 2½ years.
Association between growth velocity and SSD
The results from the final models assessing the relationship between change in height and schizophrenia-spectrum disorders are presented in Table 3. Each model controlled for all main effects; however, only relevant estimates are presented; the results for all interaction terms are presented in the online data supplement to this paper. In Table 3, the term representing the final estimate for the relationship between growth and schizophrenia spectrum disorders is labelled the ‘Effect of gender and SSD on growth, cm/year)’. Gender was included in the final estimate because patterns of growth differ between males and females (Reference KarlbergKarlberg, 1987). Furthermore, one of the goals of the analyses was to explore gender differences in the relationship between growth and schizophrenia. This estimate indicated that in SSD group growth was approximately 1 cm per year slower during early life than in the non-SSD group (B=–1.12, 95% CI −2.04 to −0.20, P=0.017), and the difference was significantly modified by gender. The difference was greater after controlling for potential confounders (B=–1.55, 95% CI −2.52 to −0.57, P=0.002). In analyses stratified by gender, in females in the SSD group growth was approximately 1 cm per year slower than in the non-SSD group (females only model; unadjusted, B=–1.03, 95% CI −1.73 to −0.33, P=0.004; adjusted, B=–1.26, 95% CI −2.00 to −0.53, P=0.001). No significant difference in growth velocity between males in the SSD and non-SSD group was observed. There was no overall or gender-specific difference in birth length between the groups or in isolated measures of height (Table 3).
Table 3 Estimated fixed effects assessing growth patterns and adult schizophrenia

| Unadjusted model | Adjusted model | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| B | 95% CI | P | B | 95% CI | P | |||||
| Early life (birth to 2½ years) | ||||||||||
| Entire sample (n=7780) | ||||||||||
| Mean height at birth, cm (intercept) | 52.59 | 52.51 to 52.66 | <0.001 | 38.76 | 38.24 to 39.28 | <0.001 | ||||
| Linear change in height, cm/year | 30.37 | 30.26 to 30.48 | <0.001 | 30.37 | 30.25 to 30.48 | <0.001 | ||||
| Quadratic change in height, cm/year2 | –6.69 | –6.74 to −6.64 | <0.001 | –6.69 | –6.74 to −6.65 | <0.001 | ||||
| Effect of SSD on birth length, cm | –0.09 | –0.77 to 0.58 | 0.784 | –0.20 | –0.64 to 0.24 | 0.375 | ||||
| Effect of SSD on linear growth, cm/year | 0.35 | –0.82 to 1.52 | 0.560 | 0.74 | –0.53 to 2.01 | 0.253 | ||||
| Effect of gender 1 and SSD on growth, cm/year | –1.12 | –2.04 to −0.20 | 0.017 | –1.55 | –2.52 to −0.57 | 0.002 | ||||
| Females only (n=3802) | ||||||||||
| Mean height at birth, cm (intercept) | 51.64 | 51.57 to 51.72 | <0.001 | 38.51 | 37.79 to 39.23 | <0.001 | ||||
| Linear change in height, cm/year | 29.54 | 29.40 to 29.68 | <0.001 | 29.54 | 29.39 to 29.68 | <0.001 | ||||
| Quadratic change in height, cm/year2 | –6.37 | –6.44 to −6.30 | <0.001 | –6.37 | –6.44 to −6.31 | <0.001 | ||||
| Effect of SSD on birth length, cm | –0.02 | –0.87 to 0.83 | 0.968 | –0.02 | –0.57 to 0.52 | 0.936 | ||||
| Effect of SSD on growth, cm/year | –1.03 | –1.73 to −0.33 | 0.004 | –1.26 | –2.00 to −0.53 | 0.001 | ||||
| Males only (n=3978) | ||||||||||
| Mean height at birth, cm (intercept) | 52.46 | 52.39 to 52.54 | <0.001 | 38.42 | 37.66 to 39.18 | <0.001 | ||||
| Linear change in height, cm/year | 30.94 | 30.79 to 31.09 | <0.001 | 30.95 | 30.79 to 31.10 | <0.001 | ||||
| Quadratic change in height, cm/year2 | –6.97 | –7.04 to −6.91 | <0.001 | –6.98 | –7.05 to −6.91 | <0.001 | ||||
| Effect of SSD on birth length, cm | –0.03 | –0.70 to 0.64 | 0.921 | –0.16 | –0.59 to 0.28 | 0.480 | ||||
| Effect of SSD on growth, cm/year | 0.06 | –0.52 to 0.64 | 0.836 | 0.26 | –0.37 to 0.89 | 0.420 | ||||
| Later childhood (2½ to 9 years) | ||||||||||
| Entire sample (n=7353) | ||||||||||
| Mean height at age 2½ years, cm (intercept) | 91.00 | 90.87 to 91.13 | <0.001 | 88.36 | 86.91 to 89.81 | <0.001 | ||||
| Linear change in height, cm/year | 7.94 | 7.89 to 7.99 | <0.001 | 7.94 | 7.89 to 7.99 | <0.001 | ||||
| Quadratic change in height, cm/year2 | –0.22 | –0.23 to −0.22 | <0.001 | –0.22 | –0.23 to −0.22 | <0.001 | ||||
| Effect of SSD on height at age 2½ years, cm | 0.53 | –0.73 to 1.80 | 0.408 | 0.17 | –1.11 to 1.46 | 0.790 | ||||
| Effect of SSD on linear growth, cm/year | –0.08 | –0.62 to 0.46 | 0.769 | –0.04 | –0.63 to 0.55 | 0.896 | ||||
| Effect of gender and SSD on growth, cm/year 1 | –0.16 | –0.61 to 0.29 | 0.478 | –0.21 | –0.70 to 0.27 | 0.389 | ||||
SSD, schizophrenia-spectrum disorders: presence of SSD=1, absence of SSD=0
1. Gender is categorised as female=1, male=0
Table 4 provides the average linear slope stratified by gender and SSD status. Dunnett's two-sided t-tests were used to compare the linear change in growth between groups stratified by gender. ‘Male non-SSD’ was used as the reference category. The results suggest that females with SSD, on average, grew 0.84 cm per year slower than males without SSD (P < 0.001); and females without SSD grew 0.23 cm per year slower than males without (P < 0.001). There was no sigificant difference in growth between males with and without SSD (Fig. 2).
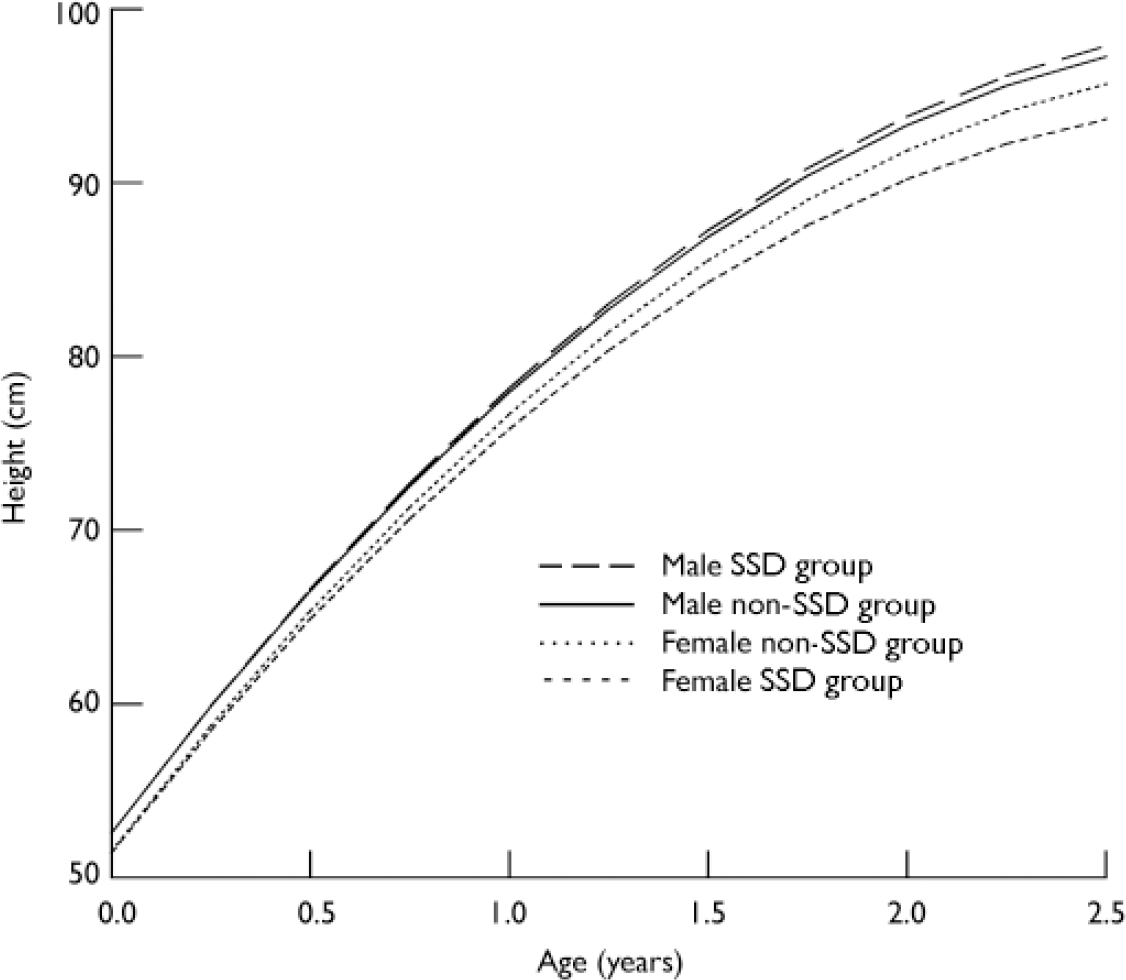
Fig. 2 Early-life growth trajectory and schizophrenia-spectrum disorders (SSD).
Table 4 Differences in linear growth stratified by gender for participants with and without schizophrenia-spectrum disorders (SSD)

| n | Linear growth, cm/year Mean (s.d.) | Difference in linear growth Mean (s.d.) | P | |
|---|---|---|---|---|
| Early life (birth to 2½ years) | ||||
| Male non-SSD | 3940 | 29.92 (0.96) | Reference | Reference |
| Female non-SSD | 3785 | 29.69 (0.91) | –0.23 (0.02) | <0.001 |
| Male SSD | 46 | 30.00 (0.81) | 0.07 (0.14) | 0.948 |
| Female SSD | 24 | 29.08 (1.26) | –0.84 (0.19) | <0.001 |
| Later childhood (2½ to 9 years) | ||||
| Male non-SSD | 3712 | 9.06 (0.45) | Reference | Reference |
| Female non-SSD | 3573 | 9.05 (0.45) | –0.02 (0.01) | 0.356 |
| Male SSD | 43 | 9.16 (0.39) | 0.10 (0.07) | 0.383 |
| Female SSD | 24 | 8.96 (0.46) | –0.10 (0.09) | 0.640 |
In contrast to early life, patterns of growth during later childhood were not significantly different between the SSD and non-SSD groups (B=–0.16, 95% CI −0.61 to 0.29, P=0.478), even after controlling for potential confounders (B=–0.21, 95% CI −0.70 to 0.28, P=0.389) (Table 3). Although we did not include the linear growth models stratified by gender for later childhood, the average linear growth estimates (Table 4) showed that girls in the SSD group continued to grow slightly more slowly, but the difference was no longer statistically significant.
In addition to assessing patterns of growth using measures of height, we also examined changes in weight and BMI. Birth weight did not differ between the SSD and non-SSD groups even after stratifying by gender. Weight gain was slightly slower in the SSD group during early life (adjusted models, B=–0.21; 95% CI=–0.64 to 0.22, P=0.342) and later childhood (B=–0.28; 95% CI–0.66 to 0.11, P=0.160), but the effect was not statistically significant. Change in BMI during infancy was comparable between the SSD and non-SSD (adjusted models, B=0.07, 95% CI −0.49 to 0.62, P=0.814). During later childhood there was a trend toward a slower change in BMI in the SSD group (adjusted models, B=–0.23, 95% CI −0.46 to 0.01, P=0.059).
DISCUSSION
This is the first study, to our knowledge, to use serial measurements and a multilevel statistical analysis to contrast patterns of early growth between people who subsequently developed a schizophrenia-spectrum disorder and those who did not. The results provide evidence that growth velocity during early life is slower in individuals who later develop schizophrenia. Furthermore, the association between delayed physical growth and adult schizophrenia was only observed in women. In contrast to previous studies, we did not find a significant difference in birth length or weight between the SSD and non-SSD groups in our cohort. We also found that isolated measures of height did not significantly differ between the two groups. Examination of patterns of growth using a series of successive rather than isolated measurements provides a more sensitive assessment of developmental abnormalities. This may explain why some studies using isolated measurements failed to find a significant difference in height between participants with schizophrenia and controls.
Our results are consistent with conclusions drawn from a previous study suggesting that the presence of delayed physical growth during early life is associated with later schizophrenia (Reference FishFish, 1959). However, that study, unlike ours, looked at a small high-risk sample, which included offspring of mentally ill parents, and thus the conclusions may not be generalisable to other populations. We have extended these findings by demonstrating this effect in a population-based cohort.
Catch-up growth and schizophrenia
Catch-up growth occurs when a period of delayed growth is followed by accelerated growth, beyond the normal rate for age (Reference Kay's and HindmarshKay's & Hindmarsh, 2006). Previous assessments of growth patterns in individuals with psychosis suggest that growth during development is atypical: these individuals are more likely to be shorter at birth but taller as adults, or taller at birth but shorter as adults (Gunnell et al, Reference Gunnell, Rasmussen and Fouskakis2003, Reference Gunnell, Harrison and Whitley2005a ). This suggests that people who develop psychosis in adulthood may experience ‘catch-up’ or ‘catch-down’ growth during development. Our study was unable to contrast the growth patterns of individuals in the SSD group who were smaller at birth and those who were larger because few participants fell into these categories. A second way to identify whether catch-up growth occurred is to examine whether individuals in the female SSD group grew more rapidly than those in the female non-SSD group during later childhood; however, this is not evident from our data. None the less, it is possible that catch-up growth might occur later in development. Future assessments of adolescent growth are necessary to explore this theory further.
Growth hormone–insulin-like growth factor axis
Our finding of slower growth velocity during early life in women with schizophrenia adds to the accumulating evidence that mechanisms responsible for the regulation of physical growth may have a role in the pathogenesis of this disorder. Although the connection between growth and schizophrenia remains to be fully elucidated, the extant literature suggests that slower growth during infancy is indicative of premorbid disruption in the growth hormone–insulin-like growth factor (GH–IGF) axis, which plays a major part in the regulation of both prenatal and postnatal growth (Reference Le Roith, Scavo and ButlerLe Roith et al, 2001). Growth during prenatal development and infancy is primarily regulated by IGF–1, as well as by insulin and nutrition, whereas growth hormone begins to play a critical part during late infancy and early childhood (Reference Karlberg, Engstrom and KarlbergKarlberg et al, 1987).
Although no previously published study has examined the association between IGF–1 levels during infancy and postnatal growth in babies born of average size, a study of adults found that lower adult IGF–1 was associated with a deceleration in growth during the first year of life, but not with birth length, weight or ponderal index (Reference Ben-Shlomo, Holly and McCarthyBen-Shlomo et al, 2003). The results from a longitudinal assessment of a large cohort suggest that IGF–1 may also be influential in regulating growth during childhood (Reference Rogers, Metcalfe and GunnellRogers et al, 2006).
The link between IGF–1 and other adult diseases such as heart disease, diabetes and hypertension has already been established (Reference Barker, Gluckman and GodfreyBarker et al, 1993; Reference Sandhu, Heald and GibsonSandhu et al, 2002). The association between birth weight and adult coronary heart disease prompted the theory that poor foetal nutrition or an insult during a critical period of development might have a long-term impact on an individual's risk of developing a number of chronic diseases later in life –referred to as the ‘foetal origins of disease hypothesis’ (Reference BarkerBarker, 1994). More recently it has been suggested that exposures operating throughout the life course might have a long-term impact on adult disease risk (Reference Ben-Shlomo and KuhBen-Shlomo & Kuh, 2002). One biological mechanism that might mediate associations of foetal and childhood growth with adult disease is perturbation of the GH–IGF axis, leading to reduced IGF–1 secretion (Reference Barker, Gluckman and GodfreyBarker et al, 1993; Reference Fall, Pandit and LawFall et al, 1995). It has been suggested that if disruption of the GH–IGF axis occurs, growth during infancy would most probably be the period of development affected (Reference Fall, Pandit and LawFall et al, 1995) because IGF–1 is more influential during infancy than during any other developmental period. Although there are alternative explanations, research linking schizophrenia and prenatal exposures such as maternal infection (Reference BrownBrown, 2006) and poor prenatal nutrition (Reference Susser and LinSusser & Lin, 1992) provides support for the foetal origins of disease hypothesis as a potential explanation for the association between slower postnatal growth and schizophrenia.
IGF-1, neurodevelopment and schizophrenia
Although the idea is speculative, it is worth considering that a disruption in IGF–1 might be a cause of abnormalities in neuro-development consistent with schizophrenia. This has been postulated to explain the observation that adults with schizophrenia are typically shorter than healthy controls, and such disruption may also be partially responsible for documented neurological abnormalities such as ventricular enlargement (Reference Gunnell and HollyGunnell & Holly, 2004).
Our findings should also be considered in light of the literature on other premorbid disturbances in schizophrenia. This body of work suggests that delays in speech and neuromotor development (Reference Jones, Rodger and MurrayJones et al, 1994) and poor intellectual functioning (Reference David, Malmberg and BrandtDavid et al, 1997) are early-life precursors of adult schizophrenia. The link between premorbid neurocognitive functioning and the GH–IGF axis is supported by a recent study of healthy children, which found a positive correlation between IGF–1 levels and performance on the Wechsler Intelligence Scale for Children (Reference Gunnell, Miller and RogersGunnell et al, 2005b ). Hence, it is possible that cognitive abnormalities and poor intellectual functioning observed prior to the development of schizophrenia might occur in part because of a dysregulation in the GH–IGF axis.
Nutrition
Nutrition also has a significant role in the regulation of growth during early life (Reference KarlbergKarlberg, 1987). Short stature and stunted growth due to malnutrition have been associated with lower scores on tests of cognitive functioning and poor educational achievement (Reference Stathis, O'Callaghan and WilliamsStathis et al, 1999; Reference StraussStrauss, 2000; Reference Berkman, Lescano and GilmanBerkman et al, 2002). In order to explore the potential role of nutrition, we assessed changes in weight and BMI during development using similar analyses to those conducted for height. No significant difference in weight or BMI between the SSD and non-SSD groups was found during early infancy. Although we did not detect any difference in weight change during later childhood, there was a trend toward slower change in BMI among participants in the SSD group. This is consistent with previous studies demonstrating associations between thinness in childhood (Reference Wahlbeck, Forsen and OsmondWahlbeck et al, 2001), low BMI in adolescence (Reference Gunnell, Harrison and WhitleyGunnell et al, 2005a ) and adult schizophrenia. Because our effect size was small, further assessments are necessary to determine whether malnourishment is responsible for the association between growth during infancy and adult schizophrenia.
Gender differences
Another noteworthy finding from our study is that gender appears to moderate the relationship between growth and schizophrenia. Women with schizophrenia-spectrum disorder grew more slowly during their early life, whereas men with the disorder did not. Although the precise neurobiological mechanisms responsible for this finding are unclear, it is known that there are gender differences not only in growth during early life, but also in secretion of and sensitivity to growth hormone and IGF–1 during development (Reference Ong, Kratzsch and KiessOng et al, 2002; Reference Geary, Pringle and RodeckGeary et al, 2003). Furthermore, gender differences in symptoms, age at onset, clinical course and treatment outcome in schizophrenia have been well documented (Reference Leung and ChueLeung & Chue, 2000). Brain imaging studies have also identified abnormal patterns of sexually dimorphic areas in schizophrenia with gender-specific differences in these abnormalities (Reference Goldstein, Seidman and O'BrienGoldstein et al, 2002). Hence, it is worth speculating that if females are more sensitive to IGF–1, they might be differentially affected by a disruption in the GH–IGF axis that could give rise to schizophrenia through neurodevelopmental mechanisms. Clearly, further research is necessary to determine the causes of gender-specific differences in the association between growth during early life and schizophrenia.
Limitations of the study
Although the results from this study are informative, a few limitations must be noted. First, since height is difficult to measure (especially during infancy), and a systematic procedure was not implemented to enhance the accuracy of measurements, it is possible that some of our measurements might have been compromised by error. Since it is unlikely that individuals in the SSD sample were more prone to measurement error than those in the non-SSD sample, this would result in non-differential misclassification error associated with height. Because the analysis included multiple measurements of each individual, it is unlikely that measurement error would produce spurious results. Second, we had to rely on maternal height as a proxy measure for target height because there were insufficient data on paternal height. It is unlikely that our inability to control properly for genetic height potential compromised the results, because the estimates from the adjusted and unadjusted models were comparable. Third, on average there were fewer measurements of height during later childhood (six measurements) than in early life (nine measurements). Fewer measurements might have limited the ability to detect a difference in patterns of growth. Linear growth models are sufficiently sensitive, however, to estimate individual growth trajectories based on two data points, so that a reduction in the number of measurements does not necessarily diminish our ability to detect a significant effect. Fourth, our analysis included a modest number in the SSD group (n=70), which might have reduced our power to detect a significant difference. Future studies of patterns of growth including a larger number of individuals with the disorder are necessary to provide more confidence in our findings.
Regardless of these methodological considerations, our study provides further support for the hypothesis that growth during early development is atypical in individuals with schizophrenia. These results indicate that growth during early life is slower in girls –but not boys –who go on to develop schizophrenia. In contrast, growth velocity during later childhood was not associated with subsequent disease status. This work also adds to the accumulating evidence for a neurodevelopmental origin of schizophrenia. Future studies assessing factors important in the regulation of growth during early life, such as growth hormone and IGF–1, will be necessary to determine the molecular and cellular mechanisms underlying the potential relationship between slowed growth and schizophrenia.
Acknowledgements
Preparation of this paper was supported by the following grants: NIMH 1R01MH 63264-01A1 (A.S.B.), a NARSAD Independent Investigator Award (A.S.B.), NICHD N01-HD-1-3334 (B. Cohn), NICHD NO1-HD-6-3258 (B. Cohn), 1R01 MH059114 (D.M.) and 2 K24 MH001699 (D.M.). We acknowledge the following individuals for their contributions to this work: Ezra S. Susser, MD, DrPH, Catherine A. Schaefer, PhD, Barbara Cohn, PhD, Michaeline A. Bresnahan, PhD, Jennie Kline, PhD, and Barbara Shine, RN. We also thank the National Institute for Child Health and Development and the Public Health Institute, Berkeley, California.









eLetters
No eLetters have been published for this article.