


Neurobiological research in schizophrenia has generally excluded patients with underlying neurological, metabolic or other medical conditions, but the study of such conditions – variably called organic psychoses, Reference Lishman1 secondary schizophrenias Reference Lewis, Hirsch and Weinberger2 or symptomatic schizophrenias Reference McKenna3 – has provided important clues and impetus for schizophrenia research. Reference Davison and Bagley4–Reference Feinstein and Ron9 With one significant exception, Reference Roberts, Done, Bruton and Crow10 few such studies have investigated the neuropathology of ‘organic psychoses’. Despite Kraepelin's concept of ‘dementia praecox’ as a frontotemporal disorder, Reference Kraepelin11 and modern neuroimaging and neuropsychological evidence of frontotemporal dysfunction in schizophrenia, Reference Lawrie and Abukmeil12,Reference Brewer, Francey, Wood, Jackson, Pantelis and Phillips13 few studies have sought an association between frontotemporal dementia and schizophrenia. Clinical diagnostic criteria for frontotemporal dementia, Reference McKhann, Albert, Grossman, Miller, Dickson and Trojanowski14,Reference Neary, Snowden, Gustafson, Passant, Stuss and Black15 reviews of its clinical features Reference Snowden, Neary and Mann16–Reference Assal and Cummings19 and frontotemporal dementia behavioural rating tools Reference Snowden, Bathgate, Varma, Blackshaw, Gibbons and Neary20 do not include psychotic symptoms. Most descriptions of schizophrenia-like psychoses in the frontotemporal dementia case literature appear to be ‘in passing’ and only a few authors have focused on this aspect of the presentation. Reference Sumi, Bird, Nochlin and Raskind21–Reference Nitrini and Rosemberg23
Our understanding of the neurobiology of frontotemporal dementia has increased dramatically in the past decade. The identification of tau, progranulin, valosin-containing protein (VCP) and CHMPB2 gene mutations in frontotemporal dementia pedigrees, Reference Hutton, Lendon, Rizzu, Baker, Froelich and Houlden24–Reference Skibinski, Parkinson, Brown, Chakrabarti, Lloyd and Hummerich29 together with the recent identification of TAR DNA-binding protein 43 (TDP-43) neuronal inclusions in the anterior horn cells of patients with motor neuron disease and the cortical neurons of frontotemporal dementia patients with and without motor neuron disease, Reference Neumann, Sampathu, Kwong, Truax, Micsenyi and Chou30 has generated renewed interest in frontotemporal dementia research.
The study reported here arose from clinical observations of patients with schizophrenia or affective psychosis, preceding any clinical diagnosis or suspicion of frontotemporal dementia. These patients, in their fourth and fifth decades, were referred on the basis of functional, cognitive or behavioural decline. Two patients, presenting before the age of 40 years, exhibited changes consistent with frontotemporal dementia associated with motor neuron disease. These clinical and clinicopathological observations led to the hypothesis that people with younger-onset frontotemporal dementia frequently present with symptoms of schizophrenia or bipolar affective disorder. The rarity of the younger-onset disorder precludes any prospective, single-centre study of such a hypothesis. We therefore investigated clinicopathological correlations in cases of younger-onset frontotemporal dementia in the National Neural Tissue Facility at the University of Melbourne, together with a detailed review of the extant case literature, to identify the characteristics of frontotemporal dementia in people that presented with schizophrenia or related psychotic disorders.
Method
Clinicopathological case series
The National Neural Tissue Facility (NNTF) is a repository of postmortem specimens representing a number of neurodegenerative and psychiatric illnesses, including cases of frontotemporal dementia (n=72). Patients who had presented with symptom onset prior to the age of 60 years were identified (n=17) and their files were examined. Demographic information (gender, age at death, age at presentation, contact details for next of kin and treating doctors) was available from files kept by the NNTF. Information regarding symptoms at clinical presentation, investigations, the course of illness, treatments and family history was obtained by an experienced neuropsychiatrist (D.V.) through a variety of sources including NNTF files, interviews and communications from treating doctors, interviews with family members and examination of coroners' reports. This information was reviewed by D.V. who determined whether a psychiatric diagnosis had been made and what that diagnosis had been. For patients diagnosed with a psychotic illness, an operational criteria checklist for psychotic and affective illness (OPCRIT) was used to generate a diagnosis according to a range of diagnostic criteria including DSM–III–R, DSM–IV and ICD–10. Reference McGuffin, Farmer and Harvey31,Reference Williams, Farmer, Ackenheil, Kaufmann and McGuffin32
Neuropathology
Brains were macroscopically examined fresh, then halved sagittally. The left side was frozen in 1 cm coronal sections and the right side was placed in 20% formalin for fixation. Formalin-fixed halves were examined after 2 weeks with sections taken from the superior frontal cortex and hippocampus. Sections were stained with haematoxylin and eosin. Immunoperoxidase studies were performed using 1E8 (in house) antibody at 1: 200, rabbit polyclonal antibodies against tau at 1: 200 (A0024; Dako, Glastrup, Denmark) and ubiquitin at 1: 100 (Z0458; Dako), and TARDBP monoclonal antibody against TDP-43 at 1: 8000 (M01, clone 2E2-D3, H00023435-M01; Abnova, Taipei, Taiwan). All cases had been reported at autopsy but were reviewed by an experienced neuropathologist (C.M.) for the purposes of this study.
Diagnoses were made based on the clinical, macroscopic, histopathological and immunohistochemical features with reference to clinical and neuropathological guidelines. Reference McKhann, Albert, Grossman, Miller, Dickson and Trojanowski14,Reference Neary, Snowden, Gustafson, Passant, Stuss and Black15 Frontotemporal dementia with tau-positive inclusions and a known tau mutation was classified as frontotemporal dementia with parkinsonism associated with a tau mutation on chromosome 17 (FTDP-17). Frontotemporal dementia with tau-negative, ubiquitin-positive inclusions was classified as FTD-u; cases of FTD-u with a clinical history of motor neuron disease were classified as frontotemporal degeneration with motor neuron disease (FTD–MND), whereas frontotemporal degeneration with motor neuron disease-like inclusions but without motor neuron disease (FTD–ID) was diagnosed if there was no clinical history of motor neuron disease but abnormal TDP-43 staining. Tau-negative, ubiquitin-negative frontotemporal dementia was classified as dementia lacking distinctive histopathology (DLDH).
Literature search strategy
A Medline search undertaken on 28 April 2007 used the terms FRONTOTEMPORAL DEMENTIA or PICK'S DISEASE or FRONTAL LOBE DEMENTIA or FRONTAL LOBE DEGENERATION, searching the years 1950–2007. Based on the initial search, subsequent searches included the following frontotemporal dementia ‘equivalent’ terms: semantic dementia, progressive aphasia, frontotemporal lobar degeneration, frontotemporal degeneration, motor neuron disease dementia aphasia complex, non-Alzheimer's presenile dementia, thalamic dementia, disinhibition-dementia-parkinsonism-amyotrophy complex and hereditary dysphasic disinhibition dementia. Additional references were obtained from reference lists of identified articles and from the Alzheimer Disease and Frontotemporal Dementia (AD&FTD) Mutation Database (www.molgen.ua.ac.be/ADMutations). Abstracts of retrieved articles were screened for the presence of frontotemporal dementia case reports or case series and the full text article then retrieved. Non-English articles were not included unless the abstract contained sufficient clinical material. Abstracts of scientific meetings were excluded. Only articles specifically cited in this paper are referenced (the full reference list is available from the author).
All retrieved articles were examined by D.V. Cases were included if all of the following information was available:
-
(a) clinical, pathological or genetic diagnosis of frontotemporal dementia (or an equivalent diagnostic term);
-
(b) age at onset of illness or at first presentation;
-
(c) clinical diagnosis or symptoms at onset or presentation of the illness.
The following information was also collected when available: age at death, gender, pathology and mutation analyses. Where possible, early studies were cross-referenced with later reports of mutation analyses in the same families or individuals and any subsequently identified mutations recorded. In familial series where not all family members were genetically assessed, it was assumed that other affected family members shared the same mutation. Cases in which a diagnosis of a psychotic illness was made (schizophrenia, schizophrenia-like illness, schizophreniform psychosis, schizoaffective disorder, bipolar disorder, psychotic depression or psychotic illness not otherwise specified) were identified and then classified according to whether the diagnosis preceded or followed the diagnosis of frontotemporal dementia.
Statistical analysis
Statistical analyses were performed using SPSS for Windows version 15. For the literature review cases the following data were described: numbers of males and females, age at onset, age at presentation, age at death, diagnosis and, where available, family history or mutation analysis results. Chi-squared analyses were used to compare gender ratio, frequency of mutations and family history for cases with and without a psychotic diagnosis. Independent t-tests were used to compare age, age at onset of symptoms, duration of illness and age at death between the groups. In order to examine whether a psychotic diagnosis at onset was more common in younger patients with frontotemporal dementia, we undertook a binary logistic regression analysis with schizophrenia-like psychosis as the outcome variable and age as the covariate.
Results
Clinicopathological series
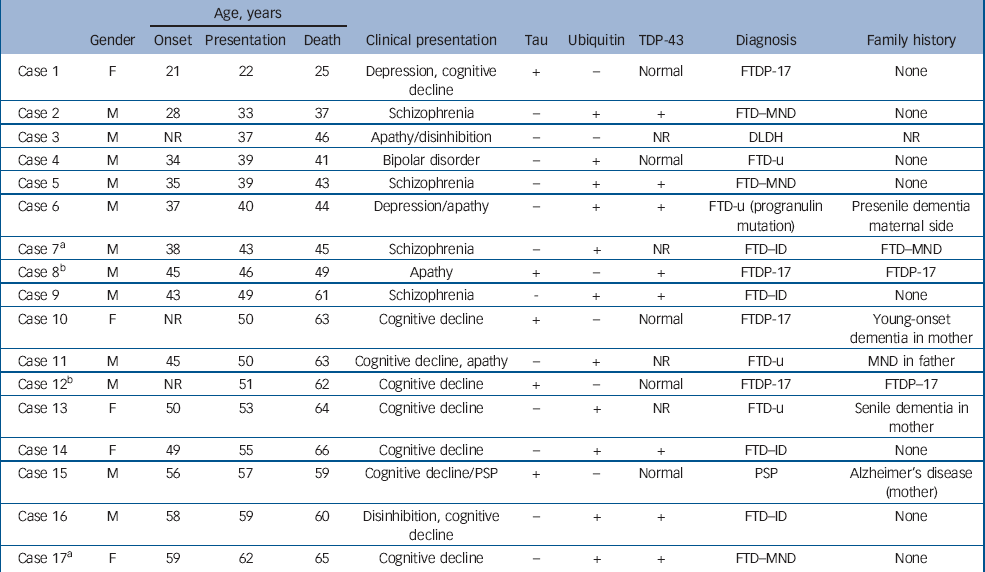
The demographic, clinical and pathological findings of the 17 cases are summarised in Tables 1 and 2. The mean ages at onset, presentation and death for the entire group were 42.7 years, 46.2 years and 52.5 years respectively. For the 5 patients with tau-positive frontotemporal dementia the respective ages were 40.7 years, 45.2 years and 51.6 years, whereas for the 11 patients with FTD-u the ages were 43.3 years, 47.5 years and 53.5 years. In cases 2, 5, 7 and 9 the patients had been diagnosed with and treated for schizophrenia; in case 4, diagnosis and treatment were for bipolar disorder. These five patients were aged 28–43 years at symptom onset (mean 35.6), 33–49 years at presentation (mean 40.6) and 37–61 years at death (mean 45.4). All had been admitted to psychiatric hospitals and diagnosed and treated by psychiatrists. The retrospective DSM–III–R, DSM–IV and ICD–10 diagnoses generated by the OPCRIT checklist were identical within each case and paralleled the original clinical diagnoses made by treating psychiatrists. Cases 2 and 5 were diagnosed with schizophrenia, case 7 with schizoaffective disorder and case 4 with bipolar disorder. Case 9 did not meet OPCRIT criteria for diagnosis but the patient had been clinically diagnosed and treated for schizophrenia. He had presented with an insidious 6-year history of amotivation, reduced energy, poor concentration and subjective memory difficulties. On assessment he exhibited loosening of associations, inappropriate affect, amotivation and disorganisation. Neurological, neuropsychological and neuroimaging investigations were initially normal; he was prescribed trifluoperazine and responded well to this treatment. The diagnosis of frontotemporal dementia was made 5 years after the initial presentation. No other patient within the neuropathological cohort met diagnostic criteria for a psychotic illness. (Detailed case histories are available from the author.)
Table 1 Young-onset frontotemporal dementia: clinicopathological series
| Age, years | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gender | Onset | Presentation | Death | Clinical presentation | Tau | Ubiquitin | TDP-43 | Diagnosis | Family history | |
| Case 1 | F | 21 | 22 | 25 | Depression, cognitive decline | + | - | Normal | FTDP-17 | None |
| Case 2 | M | 28 | 33 | 37 | Schizophrenia | - | + | + | FTD–MND | None |
| Case 3 | M | NR | 37 | 46 | Apathy/disinhibition | - | - | NR | DLDH | NR |
| Case 4 | M | 34 | 39 | 41 | Bipolar disorder | - | + | Normal | FTD-u | None |
| Case 5 | M | 35 | 39 | 43 | Schizophrenia | - | + | + | FTD–MND | None |
| Case 6 | M | 37 | 40 | 44 | Depression/apathy | - | + | + | FTD-u (progranulin mutation) | Presenile dementia maternal side |
| Case 7a | M | 38 | 43 | 45 | Schizophrenia | - | + | NR | FTD–ID | FTD–MND |
| Case 8b | M | 45 | 46 | 49 | Apathy | + | - | + | FTDP-17 | FTDP-17 |
| Case 9 | M | 43 | 49 | 61 | Schizophrenia | - | + | + | FTD–ID | None |
| Case 10 | F | NR | 50 | 63 | Cognitive decline | + | - | Normal | FTDP-17 | Young-onset dementia in mother |
| Case 11 | M | 45 | 50 | 63 | Cognitive decline, apathy | - | + | NR | FTD-u | MND in father |
| Case 12b | M | NR | 51 | 62 | Cognitive decline | + | - | Normal | FTDP-17 | FTDP-17 |
| Case 13 | F | 50 | 53 | 64 | Cognitive decline | - | + | NR | FTD-u | Senile dementia in mother |
| Case 14 | F | 49 | 55 | 66 | Cognitive decline | - | + | + | FTD–ID | None |
| Case 15 | M | 56 | 57 | 59 | Cognitive decline/PSP | + | - | Normal | PSP | Alzheimer's disease (mother) |
| Case 16 | M | 58 | 59 | 60 | Disinhibition, cognitive decline | - | + | + | FTD–ID | None |
| Case 17a | F | 59 | 62 | 65 | Cognitive decline | - | + | + | FTD–MND | None |
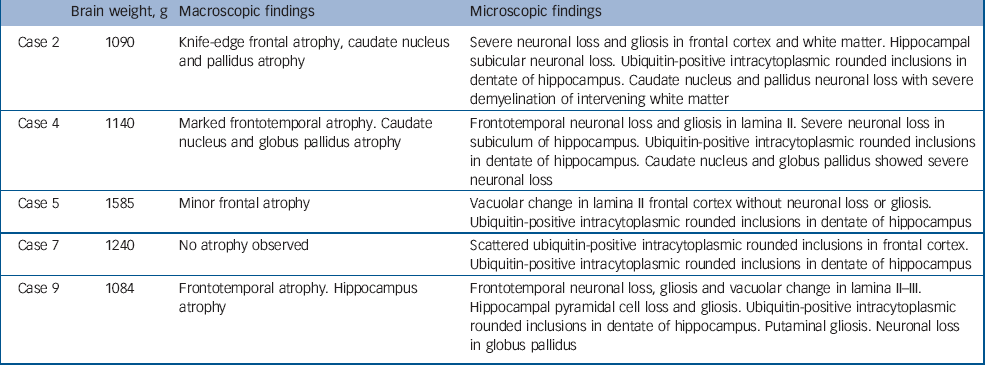
Table 2 Pathological findings in cases presenting with a psychotic illness
| Brain weight, g | Macroscopic findings | Microscopic findings | |
|---|---|---|---|
| Case 2 | 1090 | Knife-edge frontal atrophy, caudate nucleus and pallidus atrophy | Severe neuronal loss and gliosis in frontal cortex and white matter. Hippocampal subicular neuronal loss. Ubiquitin-positive intracytoplasmic rounded inclusions in dentate of hippocampus. Caudate nucleus and pallidus neuronal loss with severe demyelination of intervening white matter |
| Case 4 | 1140 | Marked frontotemporal atrophy. Caudate nucleus and globus pallidus atrophy | Frontotemporal neuronal loss and gliosis in lamina II. Severe neuronal loss in subiculum of hippocampus. Ubiquitin-positive intracytoplasmic rounded inclusions in dentate of hippocampus. Caudate nucleus and globus pallidus showed severe neuronal loss |
| Case 5 | 1585 | Minor frontal atrophy | Vacuolar change in lamina II frontal cortex without neuronal loss or gliosis. Ubiquitin-positive intracytoplasmic rounded inclusions in dentate of hippocampus |
| Case 7 | 1240 | No atrophy observed | Scattered ubiquitin-positive intracytoplasmic rounded inclusions in frontal cortex. Ubiquitin-positive intracytoplasmic rounded inclusions in dentate of hippocampus |
| Case 9 | 1084 | Frontotemporal atrophy. Hippocampus atrophy | Frontotemporal neuronal loss, gliosis and vacuolar change in lamina II–III. Hippocampal pyramidal cell loss and gliosis. Ubiquitin-positive intracytoplasmic rounded inclusions in dentate of hippocampus. Putaminal gliosis. Neuronal loss in globus pallidus |
Macroscopic pathology largely identified frontal, temporal and basal ganglia atrophy, although in two cases (cases 5 and 7) minimal macroscopic atrophy was evident. The microscopic findings revealed neuronal loss and gliosis in frontal cortex, hippocampus and basal ganglia (cases 2, 4 and 9) in cases with macroscopic atrophy, and scattered ubiquitin-positive inclusions in frontal cortex and hippocampal dentate gyrus in cases 5 and 7. All five patients had tau-negative, ubiquitin-positive inclusions in frontal and hippocampal sections consistent with a pathological diagnosis of FTD-u. Results of TDP-43 staining were available for four of the five patients (cases 2, 4, 5 and 9): three of the four exhibited a pattern of hippocampal neuronal TDP-43 staining consistent with that identified in motor neuron disease inclusions. The patient with normal TDP-43 staining was the only person with bipolar disorder within this group of five.
The 12 remaining cases comprised 5 tauopathies (4 FTDP-17 and 1 progressive supranuclear palsy) and 7 FTD-u. One of the FTD-u patients carried a progranulin mutation (case 6), having presented with depression requiring electroconvulsive therapy at age 40 years. These 12 cases presented with apathy, disinhibition, cognitive decline or personality change. All 8 patients over 50 years old at presentation (cases 10–17) had presented with cognitive decline, whereas the presentations in those 50 years old or younger were predominantly with a psychiatric diagnosis or apathy.
Literature review
The literature review identified 828 cases of frontotemporal dementia in 205 publications. From 23 publications 77 cases were excluded on the following grounds: clinical information did not meet minimum inclusion criteria (n=63) or they were members of frontotemporal dementia families who did not have frontotemporal dementia but had other diagnoses (motor neuron disease n=6, Reference Yase, Matsumoto, Azuma and Nakai33,Reference Kato, Oda, Hayashi, Kawata and Shimizu34 Alzheimer's disease n=4, Reference Lendon, Lynch, Norton, McKeel, Busfield and Craddock22 schizophrenia n=2); Reference Kertesz, Kawarai, Rogaeva, St George-Hyslop, Poorkaj and Bird35 two case reports presented patients with schizophrenia for whom frontotemporal dementia was considered but dismissed as a differential diagnosis. Reference Kimura, Inayama, Shigenobu, Kakiuchi and Tabushi36,Reference Ferrara, Freda, Massa and Carratelli37 The remaining 751 cases in 199 publications were used for the collection of data.
Cases described within the text of an article (case reports) constituted 61% of the cases, whereas 39% were reported in a tabulated form only. The majority of the 751 cases were published in the period 1990–2007 (92.0%). Single cases, 2–10 cases and 12–36 cases were reported in 44%, 49% and 7% of articles respectively. The following quantitative and qualitative variables were obtained (percentage of data in brackets): gender (92.4%, 7.6% anonymised), final diagnosis (100%), age at onset or first presentation (100%), symptoms at onset or first presentation (100%), age at death (70%), genetic mutation analysis (53%), family history of dementia (91%) and family history of psychiatric illness (73%).
Forty-six cases (6%) of frontotemporal dementia presenting with a psychotic illness were identified (online Table DS1). Overall, 30 cases (4%) were given a diagnosis of schizophrenia, schizoaffective or bipolar affective disorder; in the remaining 16 cases for which no formal diagnosis was provided by the authors, a diagnosis of psychosis has been used. The mean age at onset was 40.1 years (range 19–64) and 54% were no more than 40 years old. All but one patient (98%) had presented by the age of 60 years. For the 38 cases in which it was possible to estimate the duration of psychotic symptoms prior to the diagnosis of frontotemporal dementia, a third of patients had experienced psychotic symptoms for 2 years or less, a third for 3–10 years and a third for 11–38 years. The mean time between onset and the presentation of dementia symptoms was 10.5 years (range 0.6–38) and the mean age at death (n=28) was 57.0 years (range 30–77). There were 16 cases (35%) with one of six tau mutations and 1 case with a VCP mutation. Of the 46 cases, 8 (17%) had a clinical or pathological diagnosis of frontotemporal dementia associated with motor neuron disease. There was no difference between these latter patients and other patients with regard to age at onset of psychosis (39.8 years v. 40.2 years, t=–0.1, not significant (NS)) or age at frontotemporal dementia diagnosis (47.0 years v. 50.0 years, t=–0.6, NS), although the patients with motor neuron disease were significantly younger at death (49.3 years v. 59.1 years, t=–2.6, P=0.02). A family history of frontotemporal dementia or dementia was present in 26 cases (57%), whereas 7 (15%) had a family history of a psychotic illness.
Patients with psychosis were younger at onset (40.2 years v. 52.4 years, t=7.3, P<0.001) and experienced a longer delay between onset and subsequent presentation (11.3 years v. 2.2 years, t=13.3, P<0.001) compared with the larger frontotemporal dementia group. There was no difference in gender ratio (43% male v. 49% male, χ2=3.5, NS) or age at death (57.0 years v. 60.0 years, t=1.3, NS) between the two groups. An identified genetic mutation or a family history of dementia was less frequent in the psychosis group (57% v. 70%, χ2=3.7, P<0.05), but there was no difference in the frequency of a family history of psychiatric illness between the patients with psychosis and the other frontotemporal dementia patients (17% v. 11%, χ2=1.4, NS).
The prevalence of a diagnosis of psychosis within an age range reduced as the age range increased; for example, the prevalence of psychosis was 32.5% in patients 30 years old or younger, 20% in patients 40 years old or younger, 9.5% in patients 50 years old or younger and 6.5% in all 751 patients (Fig. 1). A diagnosis of psychosis was significantly more frequent in patients 30 years old or younger compared with patients over 30 (32.5% v. 4.2%, χ2=51.1, P<0.001), in patients 40 years old or younger compared with patients over 40 (20% v. 3.5%, χ2=47.8, P<0.001) and in patients 50 years old or younger compared with patients over 50 (9.5% v. 3.4%, χ2=12.1, P<0.01). Binary logistic regression demonstrated that younger age was highly predictive of the presence of schizophrenia-like psychosis at presentation (Wald statistic 43.2, P<0.0001, β=0.91, 95% CI 0.88–0.93).
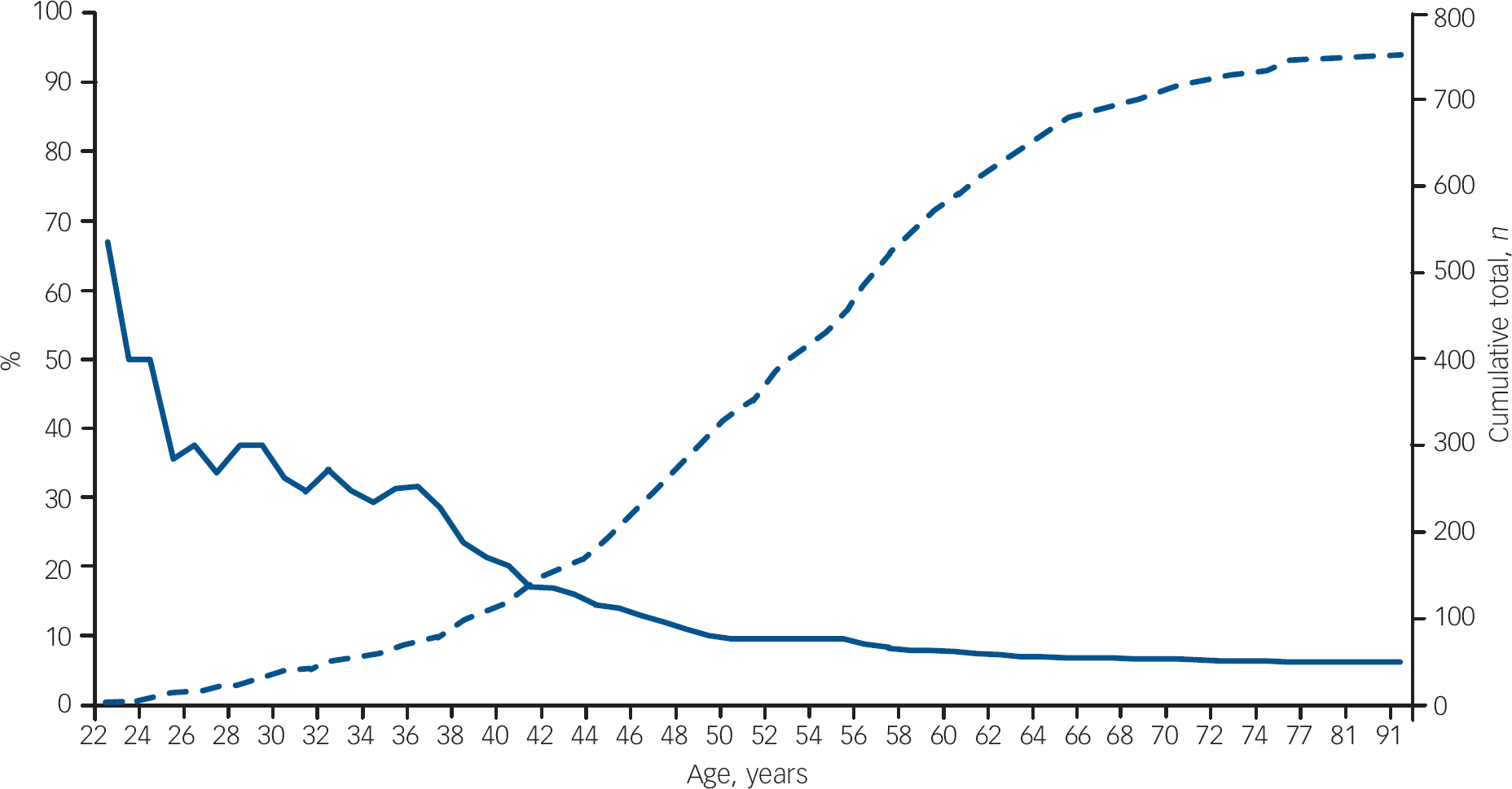
Fig. 1 Literature review: prevalence of schizophrenia-like psychosis in patients with frontotemporal dementia. The solid curve shows the percentage of patients with frontotemporal dementia presenting with schizophrenia-like psychosis at each age (left-hand axis); the dashed curve shows the cumulative number of patients with frontotemporal dementia at each age total (right-hand axis; n=751).
In 54 (7.2%) of the 751 identified cases, the patient presented with psychotic symptoms following the diagnosis of frontotemporal dementia. Psychotic symptoms during the course of illness were equally frequent in patients with frontotemporal dementia regardless of the age at illness onset and were not associated with age at onset or delay between onset and presentation.
Discussion
The neuropathological case series component of this paper describes five patients with frontotemporal dementia who presented at a young age (range 28–43 years) with a diagnosis of schizophrenia (n=4) or bipolar affective disorder and subsequently exhibited pathology consistent with FTD-u. The four patients with schizophrenia exhibited clinical or pathological (TDP-43 positive) features associated with motor neuron disease. In contrast, patients under the age of 50 years with tau-positive frontotemporal dementia presented with depression, disinhibition or apathy, and all patients with symptom onset over the age of 50 years presented with cognitive decline. Although organic ‘phenocopies’ of schizophrenia have been previously described in case reports, the strength and uniqueness of our study lies in the description of the neuropathology associated with such phenocopies and the novel finding that these patients exhibited the same pathological changes. Furthermore, in the second part of this study a comprehensive review of the frontotemporal dementia literature identified 46 cases with a diagnosis of a psychotic disorder at presentation, commonly many years before the diagnosis of frontotemporal dementia. Patients presenting at a young age were more likely to present with a psychotic illness. A third, a fifth and a tenth of all patients with frontotemporal dementia presenting at or below the ages of 30, 40 or 50 years respectively presented with a psychotic illness. Nearly all patients with a diagnosis of a psychotic disorder (98%) had presented at or before the age of 60 years, more than half were under 40 years old at presentation and 20% had a clinical or pathological diagnosis of frontotemporal dementia associated with motor neuron disease.
Previous literature regarding psychotic symptoms in frontotemporal dementia is restricted to family pedigree studies or studies that have evaluated psychotic symptoms as part of a larger survey of presenting symptoms in frontotemporal dementia. The study by Sumi et al of a family subsequently identified as carrying the tau V337M mutation identified 4 of 13 family members with a diagnosis of schizophrenia and a further 2 with a diagnosis of psychosis. Reference Sumi, Bird, Nochlin and Raskind21 This ‘schizophrenic’ phenotype was considered to be specific for this mutation, although a later study described a family that exhibited grandiose or persecutory delusions (‘megalomania’ or ‘persecution mania’) who carried the tau P301L mutation. Reference Van Swieten, Stevens, Rosso, Rizzu, Joosse and de Koning38 A high prevalence of psychosis was identified in family A in a 1973 study that did not satisfy the inclusion criteria for our literature review, Reference Sim and Bale39 but which reported that all three family members had ‘psychotic behaviour’. In a family with the VCP mutation R155C, three of six patients in family F1 had delusions and hallucinations, but further information regarding age and the relationship of the psychotic symptoms to the illness was not provided. Reference Guyant-Marechal, Laquerriere, Duyckaerts, Dumanchin, Bou and Dugny40 A study of a family with hereditary inclusion body myopathy, Paget's disease of bone and frontotemporal dementia reported that many patients had a history of auditory and visual hallucinations, but specific details regarding these patients were not provided. Reference Kovach, Waggoner, Leal, Gelber, Khardori and Levenstien41
The prevalence of psychotic symptoms at frontotemporal dementia onset has been examined in a limited way in several studies, usually as part of a larger study of neuropsychiatric symptoms at onset in frontotemporal dementia. No study was identified that specifically sought to investigate the prevalence of a diagnosis of schizophrenia or other psychotic illness during the early stages of frontotemporal dementia. In a nationwide study examining reasons for referral to geriatric psychiatric hospitals, 3% of 75 patients with frontotemporal dementia (mean age 61.6 years, range not reported, duration of illness 3.2 years) had presented with paranoid syndromes. Reference Ibach, Poljansky, Barta, Koller, Wittmann and Hajak42 In a study of early symptoms in patients with sporadic v. familial frontotemporal dementia recruited from memory clinics across two countries (n=32, mean age 53.1 years, range 33–73, duration of illness 3.6 years), 6 of 18 patients with sporadic frontotemporal dementia but none of 14 patients with familial frontotemporal dementia had a major psychiatric disorder. Reference Piguet, Brooks, Halliday, Schofield, Stanford and Kwok43 In a study of first symptoms in patients consecutively referred to a specialist dementia centre, Reference Lindau, Almkvist, Kushi, Boone, Johansson and Wahlund44 none of the 52 patients with frontotemporal dementia (mean age 64.0 years, range not reported) was identified as having psychotic symptoms. A similar absence of psychotic symptoms was identified in a study specifically examining neuropsychiatric symptoms at onset in temporal variant frontotemporal dementia (n=12, mean age 65.2 years, range not reported, duration of illness 7 years). Reference Seeley, Bauer, Miller, Gorno-Tempini, Kramer and Weiner45 In summary, these studies have tended cross-sectionally to investigate a group of older patients with frontotemporal dementia and vary markedly in the patient samples and their means of assessment. Our finding that 13.3% (100/751) of frontotemporal dementia cases were reported to have psychotic symptoms either at onset or during the course of illness falls roughly within the prevalence ranges of these studies.
Although two cases in our series (cases 5 and 7) had a family history of neurodegenerative disease, only the family history of case 7 was available in detail. Case 7's mother (case 17) had an initial clinical diagnosis of Alzheimer's disease and then subsequently developed motor neuron disease. Case 7's brother (also Case 17's son) is currently a patient of D.V. having presented with hypomania, functional decline and subtle executive dysfunction. His condition has been stable on valproate with no significant change over a 4-year period. There was a strong family history over three generations of psychiatric illness, often mania, as a prodrome to a neurological condition and dementia. This strong family history of mania is interesting given that the first diagnosis in case 7 was a schizophrenic illness with a differential of a psychotic depression, whereas the OPCRIT-generated diagnosis was of a schizoaffective disorder. In the absence of the mother's diagnosis and case 7's premature death, both brothers would have been viewed as exhibiting a major psychiatric illness without any clear clinical features to merit a diagnosis of dementia. In particular, the non-progressive, subtle, executive neuropsychological deficits identified in both would have been interpreted as consistent with their psychiatric diagnosis. The observation that case 7 was not clinically diagnosable with a dementia is significant given the lack of macroscopic brain atrophy and hippocampal pathological findings relative to the other cases, and suggests that subtle, early motor neuron disease hippocampal pathological changes may be associated with psychotic presentations rather than cognitive deficits.
Study limitations
The major limitation of both the clinicopathological case series and literature review relates to the validity of the clinical diagnoses of schizophrenia and bipolar disorder. Did these patients really have schizophrenia or bipolar disorder, was the diagnosis a ‘catch-all’ diagnosis for a young person presenting with personality change, amotivation and bizarre behaviour with or without psychotic symptoms, or were the patients mis-diagnosed? In our clinicopathological series the patients had all presented with several years of personality and behavioural changes, had all been psychiatrically assessed and had been diagnosed as having schizophrenia. They were treated by psychiatrists and received psychotropic medications resulting in clinical benefit consistent with schizophrenia. Cases 5 and 7 had a history of delusional ideation, thought disorder was a prominent feature in cases 7 and 9, and case 7 described telepathic powers. All cases exhibited symptoms considered to be negative symptoms of schizophrenia such as amotivation, personality change, apathy, reduced socialisation and insidious psychosocial decline. The use of a validated operational checklist, the OPCRIT, provided further support for the validity of these diagnoses. Current diagnostic criteria allow for a diagnosis of schizophrenia in the presence of psychosocial decline, inappropriate affect, thought disorder and disorganised behaviour. 46 The concepts of negative symptom schizophrenia and residual schizophrenia refer to a group of patients with a history of insidious psychosocial decline associated with reduced socialisation, poverty of thinking, bizarre or disorganised behaviour and thought disorder. In this context the diagnoses in cases 2, 5, 7 and 9 were justified given the information available to the psychiatrists at the time of each patient's presentation.
There are several further limitations of this study. First, there was significant variation in the quality and detail of the case reports and clinical information within tables. The primary focus of such publications was not on psychiatric aspects of frontotemporal dementia and it was not unusual to find that psychotic symptoms were reported but not discussed, or were discussed in terms of their rarity. The description of psychotic symptoms or the diagnosis of schizophrenia varied from single-word entries of ‘schizophrenia’ in a table to detailed descriptions of a patient's symptoms. Information was included from both extremes, as long as the case met the inclusion criteria outlined earlier. This approach precluded a critical review of the validity of the diagnosis or detailed differentiation between positive and negative symptoms but was considered to be necessary in order to capture as much information as was possible. A second limitation relates to the comprehensiveness of the literature review. Non-English publications, particularly from Japan, were not included, and although the Medline search terms were broad, the searches may not have revealed all published frontotemporal dementia cases, particularly cases of sporadic frontotemporal dementia. Frontotemporal dementia mutation cases were identified through the AD&FTD Mutation Database, and we were confident that all cases reported within the frontotemporal dementia mutation literature were identified and screened for inclusion criteria. Finally, it is important to acknowledge that our retrospective case-report method, which relies on clinical diagnosis not independent elucidation of diagnosis, may be inherently biased by the overreporting of psychosis or schizophrenia.
Explaining the findings: more than a frontotemporal coincidence?
Several alternative explanations for our findings are possible. A young person presenting with unusual behaviour, emotional blunting, social withdrawal, declining psychosocial functioning and subtle executive dysfunction with or without frontotemporal changes on imaging is likely to receive a psychiatric diagnosis of schizophrenia. Subsequent revision of the diagnosis to frontotemporal dementia leads to the retrospective conclusion that the patient did not have ‘real’ schizophrenia, but a schizophrenia ‘phenocopy’ or a ‘schizophrenia-like psychosis’. This explanation views our findings as a ‘coincidence’ of frontotemporal dysfunction. A second explanation is that the inverse relationship between age at onset and prevalence of schizophrenia-like presentations mirrors that described in other neurodegenerative disorders such as metachromatic leucodystrophy Reference Hyde, Ziegler and Weinberger47,Reference Walterfang, Wood, Velakoulis, Copolov and Pantelis48 and Niemann–Pick disease type C. Reference Walterfang, Fietz, Fahey, Sullivan, Leane and Lubman49 The increased rate of schizophrenia-like psychosis in early frontotemporal dementia might be the result of an interaction between neuropathology and normal central nervous system development, Reference Benes50 and might represent a non-specific, psychotic response to insult by the developing brain. According to this explanation the occurrence of a psychotic illness in younger adults with frontotemporal dementia is a non-specific effect of a neurodegenerative insult in the younger brain. This interaction may be relevant not only in terms of the developing brain but also with regard to a young adult brain which has greater neuroplastic capacity to respond to insults that cause it to age prematurely. Reference Kirkpatrick, Messias, Harvey, Fernandez-Egaz and Bowie51
The observed relationship between schizophrenia and frontotemporal dementia may be due to shared localisation of brain pathology, i.e. the occurrence of a pathological process in the same brain regions of young adults leads to a similar clinical phenotype. Indirect evidence for this can be found in the overlap of clinical, neuropsychological and neuroimaging findings in schizophrenia and frontotemporal dementia. Echopraxia, echolalia, paratonia (gegenhalten), aprosody of speech, primitive reflexes, utilisation behaviour, ‘negative’ symptoms, self-neglect, and bizarre, compulsive, impulsive and stereotyped behaviours are well recognised in both disorders. Executive dysfunction with relative preservation of visual perception and spatial skills, Reference Snowden, Neary and Mann16,Reference Pantelis, Barnes, Nelson, Tanner, Weatherley and Owen52 and deficits in social cognition, theory of mind, empathy and affect recognition have been identified in both disorders. Reference Shamay-Tsoory, Aharon-Peretz and Levkovitz53,Reference Kessels, Gerritsen, Montagne, Ackl, Diehl and Danek54 A recent study of people at risk of frontotemporal dementia identified executive and attentional dysfunction in frontotemporal dementia mutation carriers (mean age 31 years), decades before the predicted age of frontotemporal dementia onset. Reference Geschwind, Robidoux, Alarcon, Miller, Wilhelmsen and Cummings55 The pattern of neuropsychological impairment described in that study is not dissimilar to that identified in patients in the early stages of schizophrenia. Reference Pantelis, Barnes, Nelson, Tanner, Weatherley and Owen52 Frontal, temporal and hippocampal atrophy, Reference Lawrie and Abukmeil12,Reference Velakoulis, Wood, Wong, McGorry, Yung and Phillips56 and regionally specific reductions in the anterior corpus callosum Reference Kaufer, Miller, Itti, Fairbanks, Li and Fishman57,Reference Walterfang, Wood, Reutens, Wood, Chen and Velakoulis58 and anterior hippocampus, Reference Laakso, Frisoni, Kononen, Mikkonen, Beltramello and Geroldi59,Reference Velakoulis, Stuart, Wood, Smith, Brewer and Desmond60 have been described in magnetic resonance imaging studies of both frontotemporal dementia and schizophrenia. Frontal hypoperfusion on single photon emission tomography or positron emission tomography constitutes one of the imaging criteria for the diagnosis of frontotemporal dementia, Reference McKhann, Albert, Grossman, Miller, Dickson and Trojanowski14 and is also one of the most robust functional imaging findings in the schizophrenia literature in patients with chronic and first-episode illness. Reference Velakoulis and Pantelis61
Finally, it may be that a subgroup of patients who are diagnosed with schizophrenia have an insidious, slowly evolving frontotemporal dementia associated with motor neuron disease-like pathology beginning in the hippocampus. The finding that all of our clinicopathological cases with a psychotic diagnosis exhibited the same pathological changes and that these changes were evident in the hippocampus provides some support for this hypothesis, but it remains to be confirmed whether our finding is coincidental or specific. Motor neuron disease and schizophrenia have rarely been associated except in the case reports described herein and psychotic illness is not seen as an integral psychiatric feature of motor neuron disease. Reference Lishman1 Until the recent identification of TDP-43 as the protein within inclusions, the characteristic histopathological feature of frontotemporal dementia associated with motor neuron disease was of ubiquitin-positive inclusions. Such inclusions have not been identified in neuropathological studies of schizophrenia, Reference Arnold, Trojanowski, Gur, Blackwell, Han and Choi62,Reference Horton, Forsyth, Sibtain, Ball, Bruton and Royston63 and to date there has been no study examining TDP-43 in schizophrenia.
Implications of the study
Despite the strong evidence for frontotemporal deficits in schizophrenia, few modern authors have drawn parallels between schizophrenia and frontotemporal dementia. This may reflect the differences in the age at onset of the disorders, the belief that psychotic symptoms are rare in frontotemporal dementia, and the prevailing neurodevelopmental model of schizophrenia. The findings of our study may provide impetus for further exploration of the relationship between the two disorders. Although the relationship may prove to be a ‘frontotemporal coincidence’, further investigation of the schizophrenia-like psychoses associated with frontotemporal dementia may inform our understanding of the neurobiological underpinnings of schizophrenia. Finally – and importantly – there are significant clinical implications of our findings. The most common diagnosis in younger patients presenting with psychotic symptoms, personality or behavioural changes and mild executive dysfunction is of a functional psychosis such as schizophrenia. The findings of this review, however, alert clinicians to the possibility that such a presentation may represent the early stages of frontotemporal dementia. To date, psychotic symptoms have been viewed as uncommon in frontotemporal dementia and indicative of a functional diagnosis. Our findings are as relevant to neurologists assessing young patients with personality and behavioural changes as they are to psychiatrists assessing and managing patients with schizophrenia.
Acknowledgements
The authors especially thank Ms Fairlie Hinton, Ms Jane Beard, Ms Laura Leone and Mrs Tiffany Cowie for their assistance during the course of the study.





eLetters
No eLetters have been published for this article.