


Studies suggest depression in older people (commonly known as late-life depression) has a different aetiology from depression in younger adults. Reference Baldwin, Jacoby, Oppenheimer and Dening1 In particular, accumulating evidence showing that cerebrovascular disease plays an important role in late-life depression has led to the ‘vascular depression’ hypothesis. Reference Alexopoulos, Meyers, Young, Campbell, Silbersweig and Charlson2 This proposes that cerebrovascular disease and associated tissue ischaemia in key brain areas (prefrontal and subcortical grey matter) is an important cause of depression in older people. As the importance of genetic and early-life aetiological factors for major depression diminishes in later life, so neurobiological factors come to play a correspondingly larger role. Reference O'Brien, Thomas and Goodyear3 Consequently, there is evidence that depression beginning in later life, where the first episode of depression occurs after age 60 years (late-onset depression), has a larger contribution from cerebrovascular disease Reference Salloway, Malloy, Kohn, Gillard, Duffy and Rogg4 and it is therefore important to explore the role of age at depression onset within the larger group of all people with late-life depression; that is, whether older people whose illness begun earlier in life (before age 60, early-onset depression) may have different neurobiological features from those with late-onset depression. The dorsolateral prefrontal cortex has been identified as a key area in depression and previous quantitative post-mortem neuropathological studies have identified abnormalities in neurones and glia in the dorsolateral prefrontal cortex in younger adults. Reference Miguel-Hidalgo, Baucom, Dilley, Overholser, Meltzer and Stockmeier5,Reference Rajkowska, Miguel-Hidalgo, Wei, Dilley, Pittman and Meltzer6 We have reported increases in the inflammatory markers intercellular adhesion molecule-1 (ICAM-1) Reference Thomas, Ferrier, Kalaria, Woodward, Ballard and Oakley7 and vascular cell adhesion molecule-1 (VCAM-1) Reference Thomas, Ferrier, Kalaria, Davis and O'Brien8 in the microvessels of both grey and white matter of the dorsolateral prefrontal cortex in late-life depression. In autopsy studies, we have also reported an excess of ischaemic deep white matter hyperintensities in people with depression compared with controls and the great majority of these ischaemic lesions were found in the white matter of the dorsolateral prefrontal cortex, Reference Thomas, O'Brien, Davis, Ballard, Barber and Kalaria9 suggesting the inflammatory response may be as a result of cerebrovascular disease and associated ischaemia. To our knowledge, there has been only one previous three-dimensional morphometric study in the prefrontal cortex in late-life depression. Rajkowska and colleagues Reference Rajkowska, Miguel-Hidalgo, Dubey, Stockmeier and Krishnan10 estimated glial and neuronal densities in 15 people with late-life depression and 11 controls, and found no change in glial density but a significant reduction in pyramidal neuronal density, most marked in pyramidal layers 3 and 5. They did not examine neuronal volume. We examined glial and neuronal density and neuronal volume in the dorsolateral prefrontal cortex in late-life major depression. We hypothesised that people with depression would have reduced neuronal density and reduced neuronal volume as a result of tissue ischaemia in accordance with the ‘vascular depression’ model.
Method
Sample
We obtained brain tissue from the Newcastle Brain Tissue Resource from elderly people who during life had major depression and elderly comparison controls. Permission for post-mortem research had been given by the individuals and ethical approval has been granted for the use of the tissue in this study. Most of the tissue analysed was from individuals included in our previous reports Reference Thomas, Ferrier, Kalaria, Perry, Brown and O'Brien11,Reference Thomas, Ferrier, Kalaria, Davis and O'Brien8 but some tissue was from individuals new to the Newcastle Brain Tissue Resource. We reviewed the case notes for information on vascular risk factors and medication.
All tissue was from individuals who were over 60 years old; 17 individuals with DSM–IV major depression 12 and no history of any other psychiatric or neurological disorder, and 10 psychiatrically healthy comparison controls. All comparison controls were known to be capable of living independently and met the same criteria except they had never suffered a depressive episode. Individuals with depression had all received a clinical assessment and diagnosis during life by senior psychiatrists, and the Newcastle Clinicopathological committee (including senior research psychiatrists and neuropathologists) reviewed all their medical records after death together with information from the post-mortem and standardised neuropathological assessments. Diagnoses were confirmed and individuals were excluded if they showed evidence of significant cognitive impairment during life or if they met neuropathological criteria for any known cause of dementia (e.g. Alzheimer's disease, vascular dementia or dementia with Lewy bodies) or had evidence of any other neurological disorder.
Demographic, clinical and histopathological information on the study sample is summarised in Table 1. There were no significant differences between the groups in age (d.f. = 25, t = 0.324, P = 0.749), gender (χ2 = 0.318, P = 0.573), tissue pH (d.f. = 25, t = 1.539, P = 0.136), duration of tissue fixation (d.f. = 25, t = 0.453, P = 0.655), post-mortem interval (d.f. = 25, t = 0.908, P = 0.373) and brain weight (d.f. = 22, t = 0.762, P = 0.454). The causes of death were similar in the two groups, with only one individual with depression dying by suicide. The clinical features of the individuals with depression showed that in most cases they had experienced severe depressive illnesses as all except one had required in-patient care for their depression and most had received at least one course of electroconvulsive therapy. They had all received standard antidepressant treatment regimes, with selective serotonin reuptake inhibitors or tricyclic antidepressants singly or often in combination with other agents. Age at depression onset was determined from the case-note review. No controls had taken any antidepressant or mood-stabilising medication. From the case-note review, we found similar overall levels of key vascular risk factors in both groups (myocardial infarction, angina pectoris, hypertension, diabetes mellitus, atrial fibrillation, cardiac failure and smoking), as in our previous report. Reference Thomas, Ferrier, Kalaria, Perry, Brown and O'Brien11
Table 1 Demographic, histopathological and clinical information for individuals with depression and controls

| Depression group n = 17 | Control group n = 10 | |
|---|---|---|
| Age at death, years: mean (s.d.) | 76.1 (7.05) | 77.0 (7.69) |
| Gender, n | ||
| Male | 5 | 4 |
| Female | 12 | 6 |
| Histopathological information, mean (s.d.) | ||
| pH | 6.17 (0.34) | 5.96 (0.34) |
| Fixation, months | 104.35 (62.91) | 92.60 (68.93) |
| Post-mortem interval, h | 33.24 (20.33) | 26.90 (10.79) |
| Brain weight, g | 1215 (154.96) | 1165.63 (137.83) |
| Clinical features | ||
| Age at depression onset, years: mean (s.d.) | 63.65 (14.02) | |
| Depressed at death, n | 14 | |
| Required in-patient psychiatric care, n | 16 | |
| Had received electroconvulsive therapy, n | 10 | |
| Taking antidepressants at death, n | 12 | |
| Cause of death, n | ||
| Suicide | 1 | 0 |
| Pneumonia | 6 | 2 |
| Carcinoma | 2 | 4 |
| Ischaemic heart disease | 2 | 1 |
| Cardiac failure | 2 | 2 |
| Cardiac arrest | 3 | 1 |
| Renal failure | 1 | 0 |
Tissue
After death, the right hemisphere was fixed in 10% formalin. Tissue pH and brain weight were recorded for tissue from each individual and the delay from death to formalin fixation was noted. Tissue slices were obtained and sampled from pre-defined dorsolateral prefrontal cortex blocks (Brodmann areas 9 and 46). Coronal slices from each individual were selected to include Brodmann areas 9 and 46 according to a standard map. Reference Perry13 These blocks were embedded in paraffin wax and the duration of fixation of these blocks was recorded. A series of 20 30 μm sections was cut from each block and stained using cresyl fast violet. The quality of the sections and the staining was carefully checked for consistency and all slides were coded so that all analysis could be carried out blind to diagnosis. For each individual, 3 of the 20 sections were randomly sampled from these sections for morphometric analysis.
Neuronal and glial estimates
To test our hypotheses, we estimated neuronal density and volume and glial density across the whole cortex and in laminae 3 and 5. We chose these layers based on the previous evidence of neuronal pathology in the orbitofrontal cortex in late-life depression Reference Rajkowska, Miguel-Hidalgo, Dubey, Stockmeier and Krishnan10 and because we wished to avoid over-analysis by making these estimates in every cortical layer. Neurones were identified using standard criteria: that is, the presence of a nissl-stained cytoplasm, pale nucleus and single identifiable nucleolus in cells that were not spherical like glia. Pyramidal neurones were distinguished from non-pyramidal neurones by the former having a characteristic triangular morphology with a prominent apical dendrite and smaller basal dendrites. Glial cells were identified by their spherical shape, absence of nissl staining in the cytoplasm and the heterogenous arrangement of chromatin in the nucleus. The consistent application of these criteria were checked by the main rater (A.K.) working together with the senior investigator trained in stereological methodology (A.T.) and the experienced microscopist and research neuroscientist (A.O.). We viewed the sections in oil using a Zeiss Axioplan photomicroscope with a × 100 objective and numerical aperture of 1.25. The microscope was attached to a JVC colour video camera TK-C1360B (JVC UK Ltd, London, UK), a motorised x- and y-axis stage accurate to 1 μm (Optiscan ES110, Prior Scientific Instruments Ltd, Cambridge, UK) and a Heidenhain z-axis depth gauge accurate to 0.5 μm (Heidenhain GB Ltd, London, UK) to ensure accurate measurement of disector depth. We used image analysis software (Histometrix Version 5, Kinetic Imaging Ltd, Liverpool, UK) to apply the disector Reference Gundersen, Bagger, Bendtsen, Evans, Korbo and Marcussen14,Reference Sterio15 and nucleator Reference Gundersen16 methods to obtain our density and volume estimates.
Before conducting this study, we had carried out a pilot study to ensure we sampled neurones and glia evenly across the six neocortical laminae and in sufficient numbers to produce meaningful estimates. Based on our findings, we used a random sampling strategy across the cortex, with the individual laminae 3 and 5 counted separately. We conducted estimates using one disector per field and over 100 fields across the cortex and over 35 in each pyramidal layer for each section. Each disector was 64.7 μm long, 54.4 μm wide and 15 μm deep. Using the Heidenhain depth gauge, we measured the section thickness of the stained sections and found them to be a mean of 27.4 μm thick, giving a mean guard area of over 6 μm above and 6 μm below the dissector (we were aware that microtomes do not provide accurate measures of section thickness and think the sections were actually cut therefore at greater than 30 μm). There was no difference in section thickness after processing between the groups (t = 1.15, P = 0.26) and thus no evidence of differential shrinkage. The mean coefficients of error for the neuronal and glial estimates showed they were made with a high degree of precision (for glial density 2.2%; for pyramidal neurone density 3.0%; for non-pyramidal neurone density 2.4%; for pyramidal neurone volume 0.3%; for non-pyramidal neurone volume 2.5%).
Statistics
Tests for normality were conducted and unpaired, two-way Student's t-tests were used to compare the group with depression and the control group on the primary estimates and also on their basic demographic, clinical and histopathological variables. The early-onset, late-onset and control groups were compared using ANOVA in secondary analyses and we covaried as appropriate. We also used correlation coefficients to examine the effect of age at depression onset. All comparisons were carried out using SPSS (version 15.0) for Windows.
Results
Cell density and volume from the total dorsolateral prefrontal cortex
Figures 1 and 2 show box plots of the glial and neuronal cell densities respectively and Fig. 3 displays scatter plots of the cell volumes for the planned comparisons. As shown in Figs 1 and 2, there were no significant differences in glial (d.f. = 25, t = 0.089, P = 0.930), pyramidal (d.f. = 25, t = 0.129, P = 0.898) or non-pyramidal (d.f. = 25, t = 0.496, P = 0.624) density in the dorsolateral prefrontal cortex as a whole. As shown in Fig. 3, there was a significant reduction in pyramidal neuronal volume in the group with depression (d.f. = 25, t = 2.490, P = 0.020; difference in means 61.53 (95% CI for difference 10.67–112.38), which equates to a large effect size, Cohen's d = 0.91 (95% CI 0.43–1.39)). However, no such difference was found in the volume of non-pyramidal neuronal population (d.f. = 25, t = 0.620, P = 0.541). Removal of the data from the outlying individuals who had depression did not affect the significance of our finding (P = 0.022) but removal also of the two highest controls resulted in the difference becoming non-significant (P = 0.106).
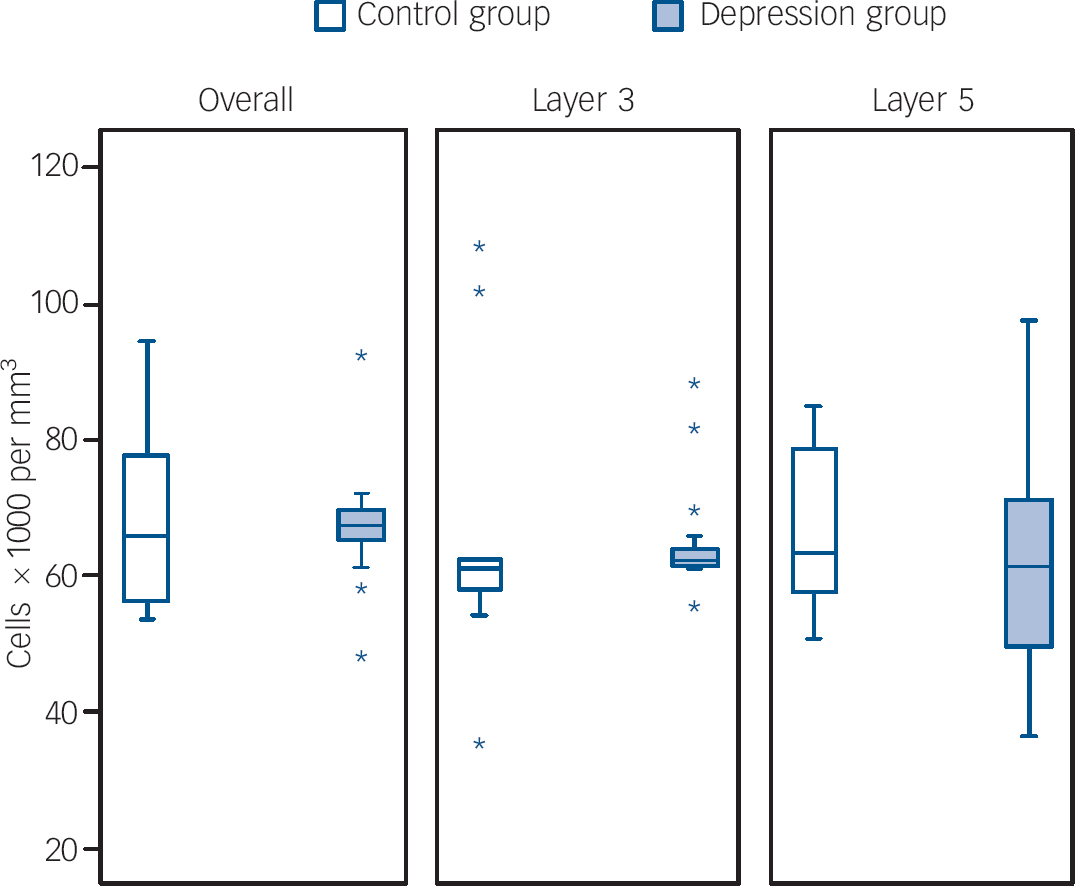
Fig. 1 Box plots of glial cell densities in layers of the dorsolateral prefrontal cortex.
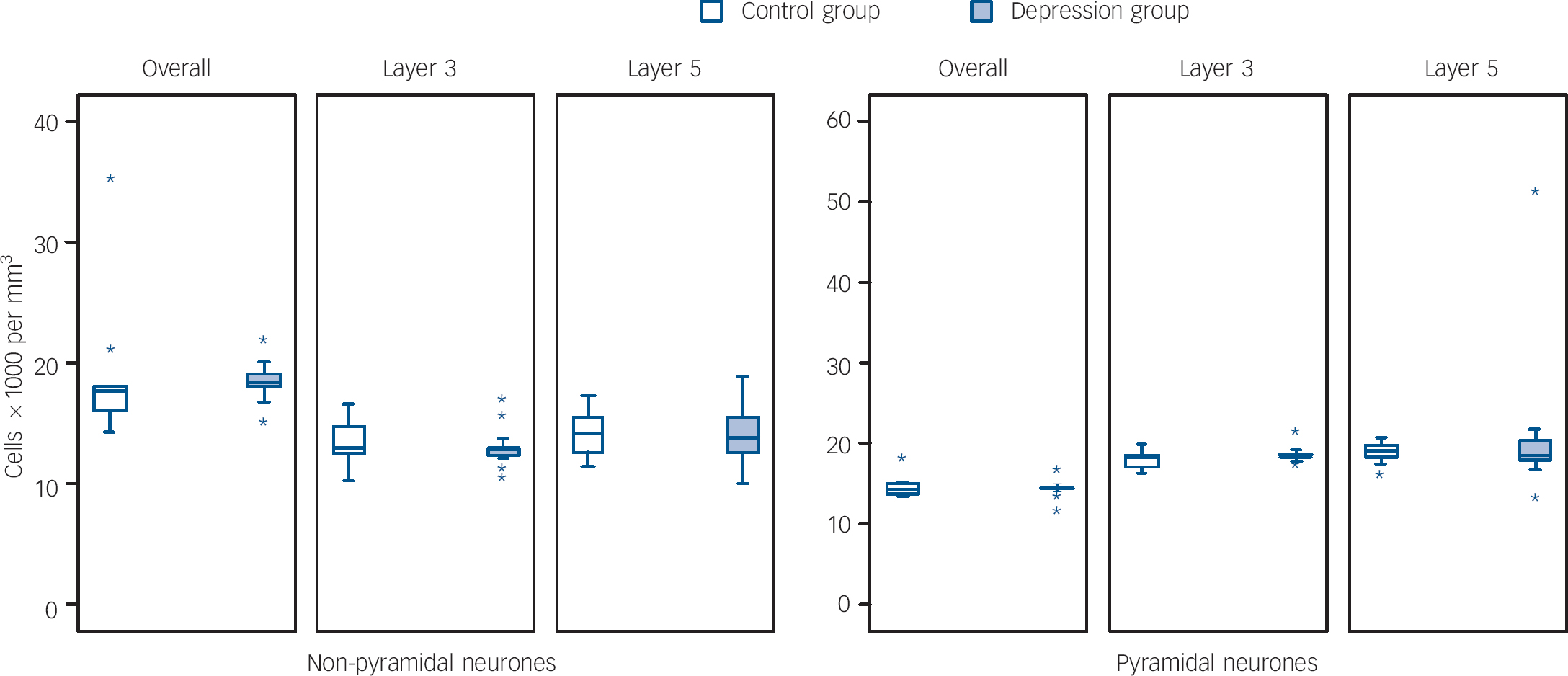
Fig. 2 Box plots of neurone densities in layers of the dorsolateral prefrontal cortex.

Fig. 3 Scatter plots of neurone volume in layers of the dorsolateral prefrontal cortex.
Cell density and volume from layers 3 and 5
Figures 1 and 2 also show the cellular density in the two pyramidal cell layers of the dorsolateral prefrontal cortex. As with the cortex as a whole, there were no differences between the two groups in layer 3 in glial (d.f. = 25, t = 0.200, P = 0.843), pyramidal (d.f. = 25, t = 0.752, P = 0.459) or non-pyramidal (d.f. = 25, t = 0.657, P = 0.517) neuronal density. Similarly, there were no group differences in layer 5 in glial (d.f. = 25, t = 0.601, P = 0.553), pyramidal (d.f. = 25, t = 0.632, P = 0.533) or non-pyramidal (d.f. = 25, t = 0.147, P = 0.884) neuronal density.
Figure 3 also shows the data for the neuronal volumes in laminae 3 and 5. In layer 3, there was a trend for pyramidal neurones to be smaller in those individuals who had depression (d.f. = 25, t = 1.963, P = 0.061; difference in means 69.72 (95% CI for difference −3.42 to 142.86)), equating to an effect size of 0.74 (95% CI 0.04–1.44), but there were no differences in non-pyramidal neurones (d.f. = 25, t = 0.839, P = 0.409). In layer 5, there was a significant reduction in the volume of the pyramidal neurones in the group with depression (d.f. = 25, t = 2.605, P = 0.015; difference in means 74.52 (95% CI for difference 15.59–133.45)), indicating a large effect (Cohen's d = 0.94 (95% CI 0.38–1.5)), but again there was no difference in the non-pyramidal neurones (d.f. = 25, t = 0.629, P = 0.535). Removal of the outlying values in layer 5 did not alter the significance of this pyramidal volume reduction (P = 0.006).
As many of the people with depression were receiving antidepressant medication when they died, it is possible our finding of reduced pyramidal neuronal volume may be a medication rather than a disease effect. To investigate this possibility, we compared the 5 individuals with depression not taking medication at death with the 12 individuals on such medication and found no significant differences in pyramidal neuronal volume between these groups in the whole cortex or in layers 3 and 5 (P>0.1 for all comparisons).
Cellular densities and volumes in early- and late-onset depression
As a secondary set of analyses, we also explored whether age at depression onset affected cellular densities and volumes. This data is shown in Figs 4, 5 and 6. Comparing groups of late-onset depression (late-onset depression, first episode of major depression at ⩾60 years n = 11), early-onset depression (n = 6), and controls (n = 10), there were no overall differences in age, gender, pH or post-mortem interval between the groups, but there was a trend level of difference in duration of tissue fixation (d.f. = 2,24, F = 2.703, P = 0.087) and so we covaried for this in all analyses. In the dorsolateral prefrontal cortex overall, there were differences in glial density between early- and late-onset depression groups (d.f. = 2,24, F = 4.086, P = 0.018), as a result of an increase in glial density in early onset of depression (early-onset depression v. controls, F = 4.769, P = 0.028; early-onset depression v. late-onset depression, F = 3.226, P = 0.070), with a smaller reduction in density in late-onset depression (late-onset depression v. controls, F = 3.870, P = 0.040). Pyramidal neuronal volume was also significantly different (d.f. = 2,24, F = 3.517, P = 0.031) as a result of reductions in neuronal volume in both early-onset depression and late-onset depression (early-onset depression v. late-onset depression, F = 2.129, P = 0.156; early-onset depression v. controls F = 3.245, P = 0.072; late-onset depression v. controls, F = 3.574, P = 0.049). There were no other differences in the whole dorsolateral prefrontal cortex and correlations controlling for fixation did not suggest a relationship between age at depression onset and glial density (r = −0.072, P = 0.792) or pyramidal volume (r = 0.163, P = 0.547) in the whole dorsolateral prefrontal cortex. There were no group differences in any density or volume estimate in lamina 3 and no correlations of any variable with age at depression onset. Findings in lamina 5 were similar to those in the whole cortex, with a significant difference in glial density (d.f. = 2,24, F = 4.553, P = 0.012) being a result of glial density increase in early-onset depression (early-onset depression v. late-onset depression, F = 4.309, P = 0.035; early-onset depression v. controls, F = 5.109, P = 0.023; late-onset depression v. controls, F = 3.256, P = 0.062). There was also a difference in the pyramidal neuronal volume (d.f. = 2,24, F = 4.633, P = 0.011), again as a result of reductions in volume in both early-onset depression and late-onset depression compared with controls (early-onset depression v. late-onset depression, F = 2.840, P = 0.092; early-onset depression v. controls, F = 4.091, P = 0.042; late-onset depression v. controls, F = 3.745, P = 0.044). In lamina 5, there was a significant difference in non-pyramidal neuronal volume (d.f. = 2,24, F = 3.144, P = 0.045) principally as a result of early-onset depression neurones being smaller than late-onset depression neurones (early-onset depression v. late-onset depression, F = 5.170, P = 0.021; early onset of depression v. controls, F = 1.491, P = 0.261; late-onset depression v. controls, F = 2.947, P = 0.078). Again, correlations showed no relationship of any estimate with age at depression onset.

Fig. 4 Box plots of glial cell densities in layers of the dorsolateral prefrontal cortex in early- and late-onset depression.
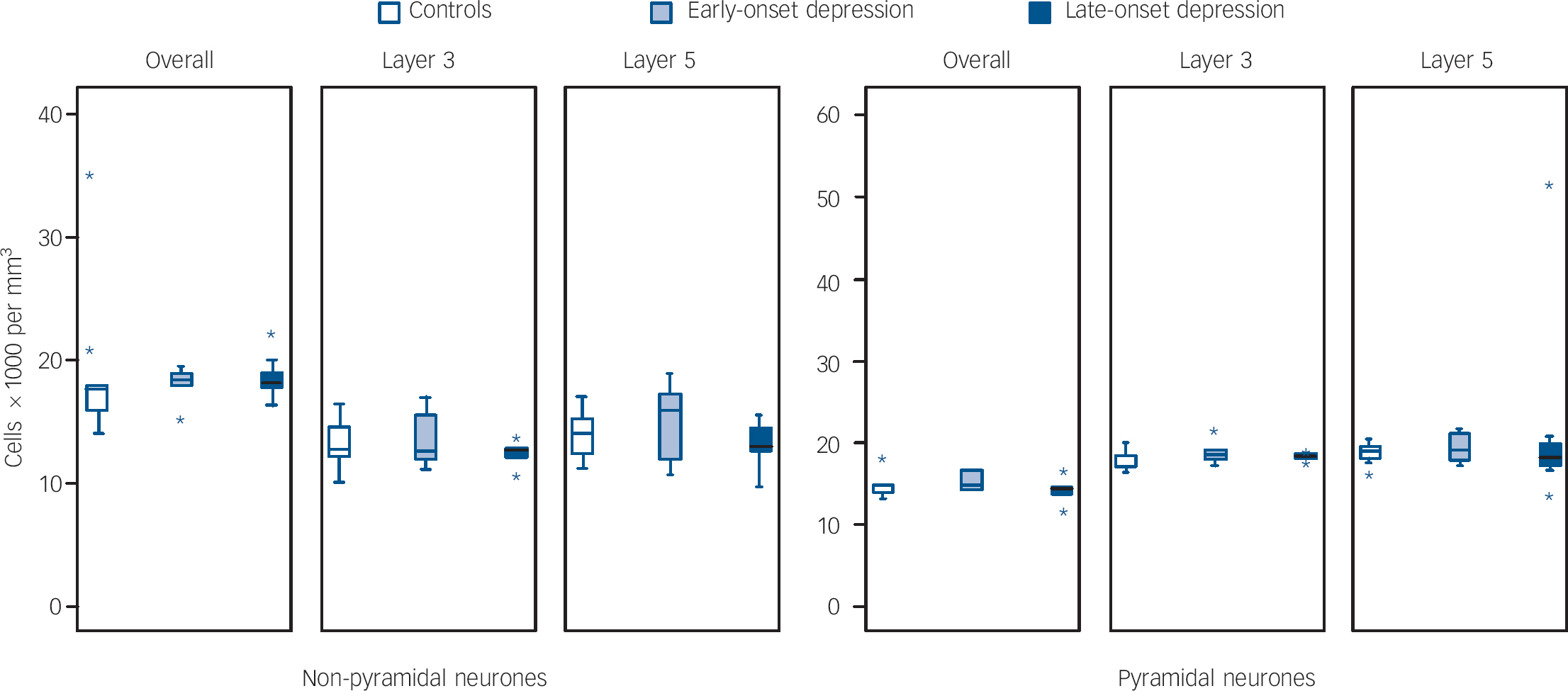
Fig. 5 Box plots of neurone densities in layers of the dorsolateral prefrontal cortex in early- and late-onset depression.
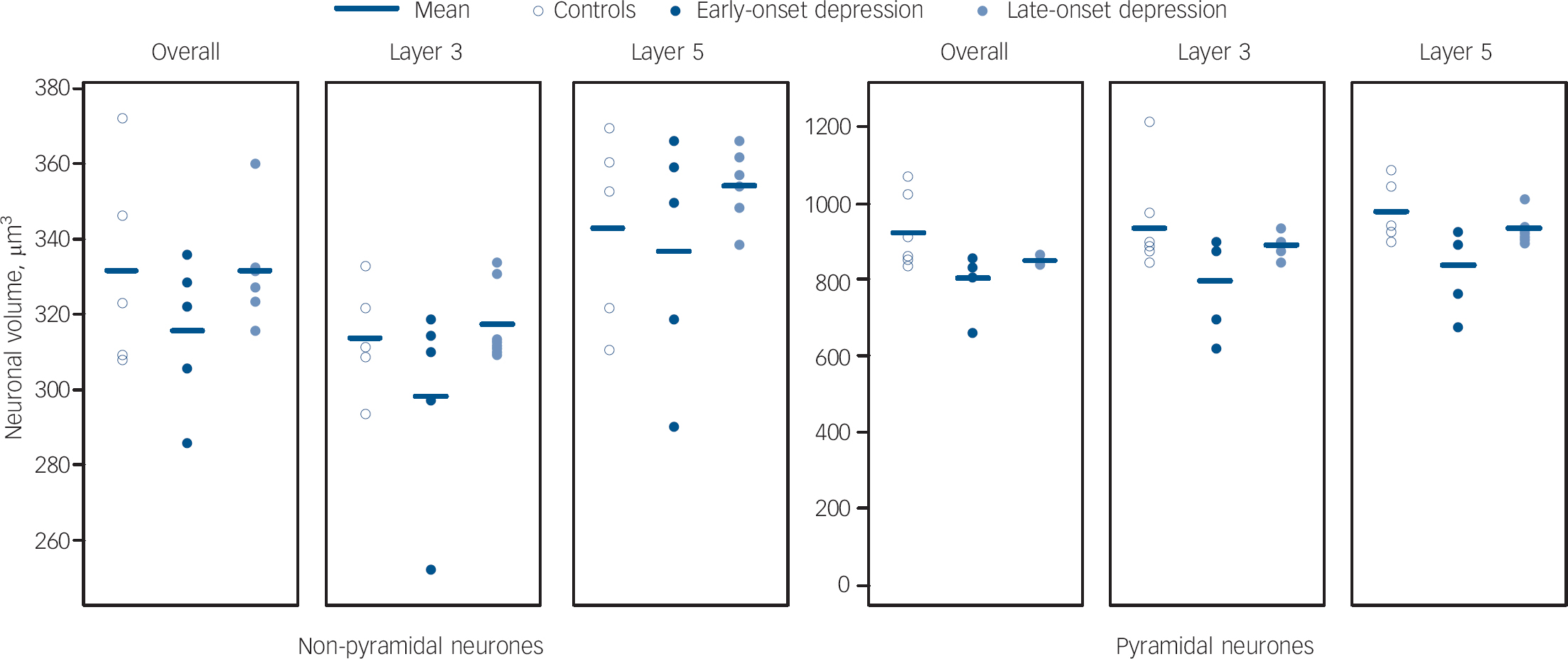
Fig. 6 Scatter plots of neurone volume in layers of the dorsolateral prefrontal cortex in early- and late-onset depression.
Discussion
We have identified pyramidal neuronal pathology, specifically reduced pyramidal cell volume, in the dorsolateral prefrontal cortex in late-life major depression. Although we did not find the reduction in pyramidal neuronal density reported by Rajkowska and colleagues in the orbitofrontal cortex, Reference Rajkowska, Miguel-Hidalgo, Dubey, Stockmeier and Krishnan10 we have also found cellular changes in the pyramidal neurone population in late-life depression. Neither our study nor the previous report from the orbitofrontal cortex found an overall glial reduction which has been consistently reported in major depression in younger adults. Reference Rajkowska, Miguel-Hidalgo, Wei, Dilley, Pittman and Meltzer6,Reference Cotter, Mackay, Landau, Kerwin and Everall17 Our finding that there was no difference in non-pyramidal cell density also agrees with Rajkouska et al and additionally we did not find any difference in non-pyramidal neuronal volume.
Prefrontal cortical pyramidal neurones are largely glutamatergic and whereas those in layer 3 are mainly association fibres projecting ipsilaterally, or less frequently contralaterally to other cortical association areas, glutamatergic neurones in layer 5 project mainly to the striatum. Reference Kaufer, Lewis, Miller and Cumming18 These glutamatergic projections from the dorsolateral prefrontal cortex synapse with GABAergic neurones in the dorsolateral aspect of the head of the caudate nucleus. These layer 5 pyramidal neurones are vulnerable to ischaemic damage as they traverse the white matter to the caudate nucleus. An increase in white matter hyperintensities is well recognised in late-life depression and in our previous report we found such lesions to be ischaemic. Reference Thomas, O'Brien, Davis, Ballard, Barber and Kalaria9 White matter hyperintensities and associated inflammation may therefore be associated with demyelination of and dysfunction in the axons of these pyramidal neurones, leading to the observed structural changes in the cortex. Since only a small proportion of layer 3 pyramidal neurones project to the caudate they would be less vulernable to such ischaemia and thus our observation that changes are more pronounced in layer 5 is consistent with this neuroanatomy.
Other explanations for our findings are possible. It is unlikely that neurodegenerative disease is a factor affecting the pyramidal neurones in our sample because we excluded all individuals with any significant degenerative disease, although a subtle effect cannot be entirely excluded. A loss of trophic support for these neurones may be present since earlier studies have implicated trophic factors, including brain-derived neurotropic factor. Reference Duman19,Reference Evans, Choudary, Neal, Li, Vawter and Tomita20 Another mechanism that could be involved is excessive hypothalamic–pituitary–adrenal axis activation, which is present in major depressive disorder in younger adults and continues into old age and tends to become more severe. Reference O'Brien, Ames, Schweitzer, Colman, Desmond and Tress21 In this model the effects of elevated steroids may lead to pyramidal neuronal atrophy, which has been proposed to explain the hippocampal atrophy observed in depression. Reference Sapolsky22
As this was necessarily a cross-sectional study, it is possible that the individuals with depression had smaller pyramidal neurones for many years. Previous investigations in younger adults have reported a reduction in neuronal volume in the anterior cingulate cortex layer 5b and 6, Reference Cotter, Mackay, Landau, Kerwin and Everall17 although this was not significant after stringent corrections for multiple comparisons, in layer 3 of the orbitofrontal cortex Reference Cotter, Hudson and Landau23 and there were suggestions of smaller neurones reported in a study of the orbitofrontal cortex and dorsolateral prefrontal cortex. Reference Rajkowska, Miguel-Hidalgo, Wei, Dilley, Pittman and Meltzer6 Thus, there may be a reduction in pyramidal volume beginning earlier in the course of depression, which becomes more marked later in the illness. Such a development would be consistent with the above vascular explanation since white matter hyperintensities are present in younger adults with depression and enlarge over time. Reference Taylor, Steffens, MacFall, McQuoid, Payne and Provenzale24
We also explored whether individuals with an early onset of their depressive illness (first episode before 60 years) differed from those with a late onset and did not find consistent evidence for such differences. Although categorical comparisons using early-onset depression and late-onset depression groups and the control group suggested some differences may be present, direct comparison of the early-onset depression and late-onset depression groups revealed only borderline differences and these were not supported by significant correlations between age at depression onset and the various estimates, either in the whole cortex or in laminae 3 and 5. Although our findings need to be interpreted with caution, in the light of the small group numbers, they are consistent with those of the previous study in late-life depression, which also did not identify significant differences between early-onset depression and late-onset depression, using the same age cut-off of 60 years. Reference Rajkowska, Miguel-Hidalgo, Dubey, Stockmeier and Krishnan10
Studies in younger adults with depression have consistently reported a reduction in glial density. Although we found no group differences in glial density, interpretation is complicated by reports of differences in glial sub-populations, which would tend to cancel each other out. We and others have reported an increase or no difference in astroglial density in late-life depression Reference Miguel-Hidalgo, Baucom, Dilley, Overholser, Meltzer and Stockmeier5,Reference Davis, Thomas, Perry, Oakley, Kalaria and O'Brien25 but a reduction in younger adults with depression Reference Miguel-Hidalgo, Baucom, Dilley, Overholser, Meltzer and Stockmeier5 and other studies report evidence of reductions in oligodendroglia in depression. Reference Aston, Jiang and Sokolov26,Reference Hamidi, Drevets and Price27
We believe the reduction in pyramidal neuronal volume in the dorsolateral prefrontal cortex is likely to be because of depressive illness itself rather than any confounding factors. As shown in Table 1, the two groups were well matched for important possible confounders (e.g. age and tissue pH), which would not therefore have biased our findings. Since the removal of outlying values for both the depression and control groups (but not that for depression alone) reduced the pyramidal volume difference to only a trend level the difference appears to be driven in part by these outliers, although the removal of two controls also further reduces the power of the analysis. Future studies need larger numbers, although this is difficult to achieve, to reduce the effect of such outliers. Another possible limitation is that although gender was not significantly different between our groups, the higher proportion of females in the depression group may have affected our findings. Inevitably, in such a study the groups were systematically different in antidepressant medication use. However, we did not find any significant difference (or trend to such a difference) in the volume of pyramidal neurones in the whole cortex or in laminae 3 and 5, making the influence of medication on our findings unlikely; this conclusion is consistent with that of the previous late-life depression study in the orbitofrontal cortex. Reference Rajkowska, Miguel-Hidalgo, Dubey, Stockmeier and Krishnan10 As our study was performed in predefined, paraffin-embedded blocks, we had to work with the limitations of such constraints that thus prevented a full stereological analysis of the dorsolateral prefrontal cortex. As it was not possible to sample the whole dorsolateral prefrontal cortex, it could be argued that a bias may occur in the results because of the precise location of the tissue obtained within the dorsolateral prefrontal cortex. Although impossible to statistically verify, we are confident that readings were obtained from a similar caudal position that did not differ between the groups.
In conclusion, consistent with the only previous study in late-life depression we have found further evidence of pyramidal neuronal changes that were more marked in layer 5 of the dorsolateral prefrontal cortex and that were not associated with any changes in the overall glial population or in non-pyramidal neurones. These findings could be because of the influence of cerebrovascular disease, especially that affecting the white matter which damages the axons of the pyramidal neurones as they project to the caudate nucleus.








eLetters
No eLetters have been published for this article.