


The Kraepelinian dichotomy – the broad division of major mood and psychotic illness of adulthood into schizophrenia and ‘manic–depressive’ (bipolar) illness – has been enshrined in Western psychiatry for over a century and continues to influence clinical practice, research and public perceptions of mental illness. Nearly 5 years ago, we published an editorial Reference Craddock and Owen1 in which we summarised emerging evidence undermining the dichotomy, and argued that continued adherence to this approach is hampering research and clinical care. Since then, a substantial and increasingly convergent body of evidence has accrued from genetic studies that supports and refines this view. Here, we summarise these findings and their implications for clinical psychiatry and illness classification.
Findings inconsistent with the Kraepelinian dichotomy
Several studies have been published in the past 5 years that provide compelling evidence that genetic susceptibility and, by implication, elements of the underlying pathogenetic mechanisms are shared between bipolar disorder and schizophrenia. Although most of this new evidence has come from large-scale molecular genetic analyses, the molecular findings are fully supported by a large family study of the two disorders. The recent evidence also points to the need to reconsider the relationship of mood and psychotic illness with other neuropsychiatric phenotypes such as autism. Key findings include the largest family study of the two disorders ever conducted that shows overlap in genetic susceptibility across bipolar disorder and schizophrenia. This study included over 2 million nuclear families identified from Swedish population and hospital discharge registers, and showed that there are increased risks of both schizophrenia and bipolar disorder in first-degree relatives of probands with either disorder. Moreover, there is evidence from half-siblings and adopted-away relatives that this is due substantially to genetic factors. Reference Lichtenstein, Yip, Björk, Pawitan, Cannon and Sullivan2,Reference Owen and Craddock3
Additionally, genome-wide association studies (GWAS) have demonstrated the existence of common DNA variants (single nucleotide polymorphisms) that influence risk of both schizophrenia and bipolar disorder. There is direct molecular genetic support for a substantial genetic overlap between schizophrenia and bipolar disorder from the recent large-scale GWAS of bipolar disorder and schizophrenia in which thousands of individuals have been studied for hundreds of thousands of common DNA variants spread across the genome. This evidence includes analyses of specific risk loci, including ZNF804A, initially identified as a risk gene for schizophrenia, Reference O'Donovan, Craddock, Norton, Williams, Peirce and Moskvina4 and CACNA1C, Reference Green, Grozeva, Jones, Jones, Kirov and Caesar5 which was strongly implicated initially in GWAS of bipolar disorder. Reference Ferreira, O'Donovan, Meng, Jones, Ruderfer and Jones6 It is of interest that there is evidence that variation at CACNA1C also influences risk of recurrent unipolar depression. Reference Green, Grozeva, Jones, Jones, Kirov and Caesar5 More broadly, there is evidence for overlap in the identity of genes showing gene-wide association signals in GWAS of schizophrenia and bipolar disorder. Reference Moskvina, Craddock, Holmans, Nikolov, Pahwa and Green7 Perhaps most compellingly, there is strong evidence that the aggregate polygenic contribution of many alleles of small effect to susceptibility for schizophrenia also influences susceptibility to bipolar disorder. 8
Although a great deal of work remains to be undertaken in delineating the pathogenically relevant DNA variants and the biological mechanisms by which they influence disease risk, the studies described above indicate that schizophrenia and bipolar disorder (and recurrent depression) do not ‘breed true’, but have an overlap in genetic risk and are therefore likely to share some aspects of pathogenesis. This does not equate to a simple dichotomous notion of completely distinct and unrelated disease categories and allows us to reject the traditional, simplistic view of the dichotomy. We think this is unlikely to surprise clinicians, who frequently have to decide how to categorise and treat individuals with a mixture of prominent mood and psychotic symptoms.
Findings indicating the need to reconsider the interface between psychosis and autism
It has recently been recognised that structural genomic variants of small or modest size (copy number variants (CNVs) of stretches of DNA of 1000 base pairs or more) are a common cause of genetic variation in humans, Reference McCarroll and Altshuler9 and such variants have been reported in neuropsychiatric phenotypes, including autism, ‘mental retardation’ (intellectual disability) and schizophrenia. Reference Weiss, Shen, Korn, Arking, Miller and Fossdal10–Reference Pagnamenta, Wing, Akha, Knight, Bölte and Schmötzer15 The overall ‘load’ of CNVs has been shown to be greater in individuals with schizophrenia compared with controls, and there is convincing evidence that a number of specific rare CNVs (<1% population minor allele frequency) confer risk of schizophrenia, particularly those at 22q11 (the velocardiofacial syndrome deletion), 1q21.1, 15q13.3 and 15q11.2, Reference Stefansson, Rujescu, Cichon, Pietiläinen, Ingason and Steinberg12–Reference Kirov, Grozeva, Norton, Ivanov, Mantripragada and Holmans14 as well as deletions of the gene encoding the synaptic neural adhesion molecule, neurexin 1. Reference Kirov, Rujescu, Ingason, Collier, O'Donovan and Owen16 The specific CNVs associated with risk of schizophrenia also confer risk of multiple neuropsychiatric phenotypes, including autism and mental retardation. Reference Burbach and van der Zwaag17 This indicates an overlap of genetic susceptibility and pathogenesis across the categories of schizophrenia, autism and other neurodevelopmental disorders and challenges the view that these are completely unrelated diagnostic entities.
Findings suggesting that bipolar disorder and schizophrenia do not have a single underlying cause and are not the same clinical entity
Although we can reject a simple model of separate, unrelated disease categories, the data do not support a model of a single-disease category that is undifferentiated with respect to the relationship between clinical expression and genetic susceptibility, and, hence, underlying biological mechanisms. For example, the same large family study Reference Lichtenstein, Yip, Björk, Pawitan, Cannon and Sullivan2 that demonstrated a substantial overlap in genetic susceptibility to bipolar disorder and schizophrenia also provided clear evidence for the existence of non-shared genetic risk factors. These findings are fully consistent with earlier genetic data suggesting that there are relatively specific as well as shared susceptibility genes. Reference Craddock and Owen1 Recent studies suggest that some of this specificity might be due to structural genomic variation (CNVs). Although there is emerging evidence that CNVs have some influence on the risk of bipolar disorder, Reference Zhang, Cheng, Qian, Alliey-Rodriguez, Kelsoe and Greenwood18,Reference McCarthy, Makarov, Kirov, Addington, McClellan and Yoon19 they appear to contribute less to the susceptibility to bipolar disorder than to schizophrenia (to date, variants influencing bipolar disorder seem to be smaller, less likely to be deletions, and have smaller effect sizes). Reference McCarthy, Makarov, Kirov, Addington, McClellan and Yoon19,Reference Grozeva, Kirov and Ivanov20 Under the assumption that bigger structural genomic variants, particularly involving DNA loss, are more likely to affect brain development, we note that these findings are consistent with the view that schizophrenia has a stronger neurodevelopmental component than bipolar disorder Reference Murray, Sham, van Os, Zanelli, Cannon and McDonald21 and suggest that it lies on a gradient of decreasing neurodevelopmental impairment between syndromes such as mental retardation and autism on one hand, and bipolar disorder on the other (Fig. 1).
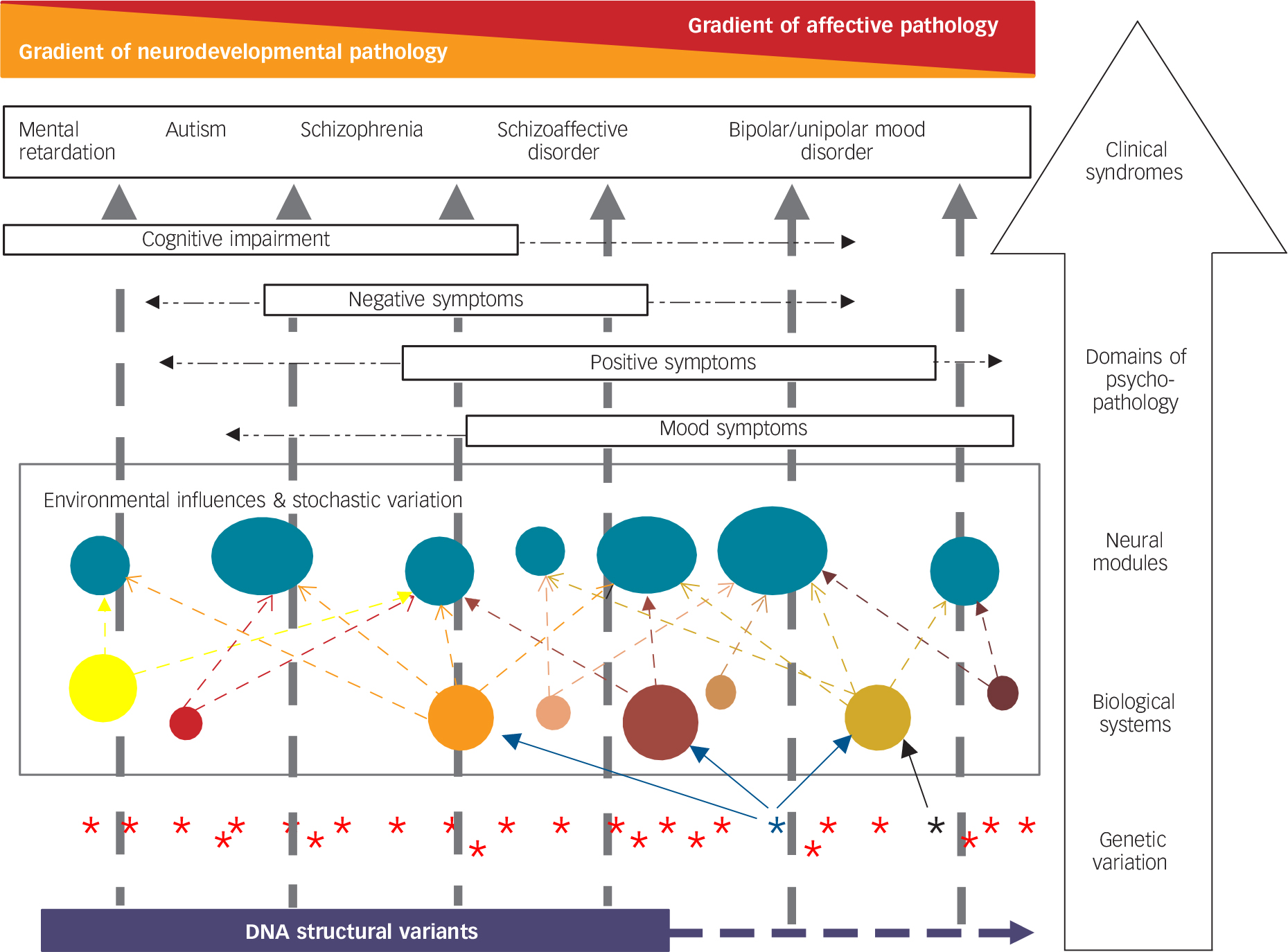
Fig. 1 Hypothesised model of the complex relationship between biological variation and some major forms of psychopathology.
This is a simplified model of a highly complex set of relationships between genotype and clinical phenotype. Starting at the level of genetic variation (lowest tier in figure), we have represented DNA structural variation (in purple) as contributing particularly to neurodevelopmental disorders and associated particularly with enduring cognitive and functional impairment. Single gene variants, of which there are many, are shown as asterisks. In general, even single base-pair changes in a gene may influence multiple biological systems because genes typically have multiple functions and produce proteins that interact with multiple other proteins. For simplicity, we have shown only an example of a variant that influences three biological systems (blue asterisk and arrows) and another that influences only one system (black asterisk and arrow). Variation in the relevant biological systems is influenced by genotype at many genetic loci and by environmental exposures/experiences both historically during development and currently to influence the dynamic state of the systems. The relevant biological systems influence the neural modules that comprise the key relevant functional elements of the brain (shown as solid turquoise circles). Typically, multiple biological systems influence each neural module. The (abnormal) functioning of the neural modules together influences the domains of psychopathology experienced and ultimately the clinical syndromes. We have ordered some important clinical syndromes along a single major axis with a gradient of decreasing proportional neurodevelopmental contribution to causation and reciprocal increasing gradient of proportion of episodic affective disturbance (we use the term ‘mental retardation’ in the diagram because it is understood internationally, but recognise that the terms intellectual disability and learning disability are commonly used in the UK). The single axis is a simplifying device – there is substantial individual variation and it is recognised that, for example, it is not uncommon for individuals diagnosed with autism to experience substantial mood pathology. Key features of the model are described within the text.
Data suggesting a degree of specificity between pathophysiology and phenotype come from work at the interface of the traditional dichotomous categories. Cases with a rich mix of clinical features of bipolar mood episodes and the psychotic symptoms typical of schizophrenia (a broadly defined schizoaffective illness) may be particularly useful for genetic studies, Reference Hamshere, Green, Jones, Jones, Moskvina and Kirov22 and there is evidence that variation within genes encoding gamma-aminobutyric acid (A) receptor subunit genes may predispose relatively specifically to such mixed mood–psychosis clinical pictures. Reference Craddock, Jones, Jones, Kirov, Green and Grozeva23 Although continued reliance on the relatively narrow DSM–IV and ICD–10 definitions of schizoaffective disorder would appear to be untenable, particularly on the grounds of poor reliability, Reference Craddock, O'Donovan and Owen24,Reference Heckers25 this clinical entity merits explicit recognition in order to explore this possibility further. We note that the abolition of a schizoaffective category from revisions to current classifications, as advocated by some (e.g. Heckers Reference Heckers25 ), would risk further reinforcing a completely inappropriate dichotomous view.
Moving towards more biologically plausible and clinically useful models of psychosis
The main clinical aims of diagnosis include the optimisation of treatments and allowing useful prognostic statements to be made. The history of medicine suggests that therapeutic and prognostic decision-making are usually facilitated, often greatly, as classifications move closer to the underlying biological mechanisms. For this reason, it is desirable to move towards a classification that maps the expression of illness onto the underlying biological systems. It is not yet clear whether this will be most usefully achieved by using multiple overlapping ‘categorical’ domains of psychopathology or multiple dimensions. Reference Craddock and Owen1,Reference Craddock and Owen26,Reference van Os and Kapur27 Clinicians benefit from the simplest, most user-friendly model that is clinically useful. Of course, the traditional dichotomy is simple and this perhaps explains its persistence despite increasingly questioned clinical usefulness. Reference Craddock and Owen1 In our 2005 editorial we suggested that recent evidence made it necessary to consider a mood–psychosis clinical dimension with at least three possible overlapping broad domains of psychopathology (‘prototype schizophrenia’, ‘schizoaffective’ and ‘prototype bipolar’). More recent genetic data are broadly consistent with such a model. However, these newer data also point to the need to consider a broader clinical spectrum that includes also autism and mental retardation/cognitive impairment at one end and non-psychotic mood disorder at the other.
A simple model of a complex set of relationships
If models of illness are to map onto the underlying functional systems of the brain, it is obviously essential to take close account of normal as well as abnormal brain function. The brain is a highly complex structure in which high levels of anatomical and functional connectivity occur at many levels. Reference Bullmore and Sporns28,Reference Zielasek and Gaebel29 Plasticity occurs at all stages of development and environmental influences cause important short- and long-term effects on brain function. Psychiatry must strive to integrate evidence from various levels of neuroscientific investigation – including molecular biology, cognitive neuroscience and affective neuroscience – to move towards a coherent understanding of psychiatric illness that can appropriately use models from each area of inquiry for the benefit of patients.
Here, we present a simple uniaxial model for the relationship between several traditional diagnostic entities that is broadly consistent with current genetic data (Fig. 1). The aim is to aid thinking, guide future work and stimulate debate. Key features of the model include:
-
(a) using dimensions/continua to conceptualise the major clinical syndromes;
-
(b) broad organisation along a major axis according to a gradient of increasing proportion of neurodevelopmental contribution to illness in one direction and increasing proportion of episodic affective contribution in the opposite direction;
-
(c) recognising that multiple domains/dimensions of psychopathology contribute to the major clinical syndromes in varying proportions and that these may relate more closely to dysfunctional brain systems;
-
(d) recognising that the states of relevant brain systems depend crucially on environmental influences (both developmentally and dynamically);
-
(e) recognising the complexity, interdependence and modular nature of brain systems (where modules are functionally discernible, not necessarily temporally or spatially stable subunits, which are interconnected in complex, often multilayered networks of neuronal circuits); Reference Zielasek and Gaebel29
-
(f) recognising that we are not dealing with a one-to-one relationship (i.e. the concept of ‘a gene for schizophrenia’ or even ‘a gene for auditory hallucinations’ is not plausible), Reference Kendler30 but rather sets of many one-to-one and/or one-to-many relationships.
Future challenges
We have entered a period of unprecedentedly rapid progress in understanding mental illness. While ensuring that the needs of our patients are at the forefront of thinking and planning, we need to prepare ourselves to move towards more complex and biologically plausible models of illness rather than clinging on to the biology-free models based on clinical empiricism that have been the tradition of psychiatry.
Although current data suggest that there is some degree of genetic specificity at the level of clinical symptoms, it is an empirical question as to what degree of specificity it will be possible to delineate and which specific domains (or dimensions or syndromes) of psychopathology will be most useful to recognise and measure. Reference Craddock, Kendler, Neale, Nurnberger and Purcell31 There is a pressing need to characterise the neurocognitive disturbances that underlie the major domains of psychopathology if we wish to develop a more refined taxonomy of mental disorders Reference Zielasek and Gaebel29 as well as better entities for genetic and other aetiological studies. It is to be hoped that more fundamental phenotypes might emerge from studies of the biological systems implicated by genetic and other biological findings.
A combination of these top-down and bottom-up approaches might ultimately allow us to trace the links between genotype and phenotype. These efforts will require greater integration between different research modalities, including genetics, psychopathology, and cognitive and affective neuroscience, together with insights from systems biology. This should be complemented by consideration of social and other relevant environmental variables, and include a developmental perspective. There is also a need for more longitudinal studies; it seems inconceivable that future taxonomies will not address the considerable variations in course and outcome seen in the clinic. This will require major investment in research, closer cooperation between different research paradigms and a movement away from schools of thought that are often held with ideological zeal.
Conclusion
At a time of transition it will be necessary to be open-minded and flexible. Care must be taken to ensure that the diagnostic entities (be they dimensions, categories or a mixture) are based on solid data, are usable and have proven clinical utility. Inevitably, research must move faster and be willing to explore a wide range of options unconstrained by current diagnostic categories. Clinical practice must expect a slower pace of change and a longer transitional period. Efforts to reformulate DSM–V and ICD–11 are currently underway. In our view, the most pragmatic solution to current needs is to encourage the careful measurement and reappraisal of psychopathology by using dimensional measures of key domains of psychopathology which can sit alongside the use of categories. The resulting detailed clinical diagnostic evaluation will allow the efficacy of current and future treatments to be monitored in individual cases and better serve research into aetiology, classification and treatment. Thus, it is likely to be a while yet before the traditional dichotomous prototypes will make an exit from official classifications, even though evidence and thinking are moving on.
At the time Emil Kraepelin introduced his dichotomy based on longitudinal course there were no effective treatments. At the end of the 19th century, it was logical to use a simple diagnostic approach that offered reasonable prognostic validity. At the beginning of the 21st century, we must set our sights higher.
Funding
The authors' work on the genetics of psychosis and mood disorders is funded through grants from the Wellcome Trust, MRC, NIH and Stanley Medical Research Institute.
Acknowledgements
We are members of the MRC Centre for Neuropsychiatric Genetics and Genomics in Cardiff University. M.J.O. is a member of the DSM–V Work Group on Psychotic Disorders. The opinions expressed in this article do not necessarily reflect the consensus of the DSM–V Work Group or Task Force. We are indebted to all the participants in our studies.


eLetters
No eLetters have been published for this article.