


Copy number variants (CNVs) are chromosomal rearrangements involving large segments of DNA (from 1000 and up to several million base pairs in length) that can be deleted, duplicated, inverted or translocated. A number of pathogenic CNVs are known to cause clinically recognisable syndromes, such as Williams-Beuren syndrome (WBS), Angelman/Prader-Willi syndrome (AS/PWS) and velocardiofacial syndrome (VCFS). Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein1 Some CNVs are associated with a highly variable phenotype, which often includes both developmental and neuropsychiatric disorders. Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein1 For example, 22q11.2 deletions, the first CNV to be associated with schizophrenia, Reference Karayiorgou, Morris, Morrow, Shprintzen, Goldberg and Borrow2 also causes VCFS, which is characterised by intellectual disability and a number of medical problems such as palatal and skeletal anomalies and cardiac defects. Reference Driscoll, Salvin, Sellinger, Budarf, McDonald-McGinn and Zackai3,Reference Shprintzen, Goldberg, Lewin, Sidoti, Berkman and Argamaso4 Several large and rare CNVs have now been implicated in the aetiology of schizophrenia, Reference Grozeva, Conrad, Barnes, Hurles, Owen and O'Donovan5-Reference Rujescu, Ingason, Cichon, Pietiläinen, Barnes and Toulopoulou13 reviewed by Malhotra & Sebat. Reference Malhotra and Sebat14 They have been shown to substantially increase risk for the development of the disorder, from twofold to over 60-fold. Reference Malhotra and Sebat14 Most of them have been shown to also increase risk for other disorders, such as autism spectrum disorders, intellectual deficit, developmental delay and epilepsy. Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein1,Reference Malhotra and Sebat14,Reference Kirov, Rees, Walters, Escott-Price, Georgieva and Richards15
As some of the CNVs are very rare (found in less than 1 in 1000 patients), notwithstanding the relatively large data-sets examined so far (ranging between ∼5000 and 14 000 people with schizophrenia), it is not clear that all those that have been implicated are true risk factors for the disorder. Thus, for nine of the loci that have received the strongest support in the literature, Reference Malhotra and Sebat14 fewer than 15 observations have been made in people with schizophrenia. These nine loci are: duplications at 1q21.1, Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 the WBS region, Reference Mulle, Pulver, McGrath, Wolyniec, Dodd and Cutler16 the AS/PWS region, Reference Ingason, Kirov, Giegling, Hansen, Isles and Jakobsen6 at VIPR2, Reference Levinson, Duan, Oh, Wang, Sanders and Shi10,Reference Vacic, McCarthy, Malhotra, Murray, Chou and Peoples17 at 16p13.11 Reference Ingason, Rujescu, Cichon, Sigurdsson, Sigmundsson and Pietilainen7 and deletions at 3q29, Reference Levinson, Duan, Oh, Wang, Sanders and Shi10,Reference Mulle, Dodd, McGrath, Wolyniec, Mitchell and Shetty12 distal 16p11.2, Reference Guha, Rees, Darvasi, Ivanov, Ikeda and Bergen18 17q12 Reference Moreno-De-Luca, Mulle, Kaminsky, Sanders, Myers and Adam19 and 17p12. Reference Kirov, Grozeva, Norton, Ivanov, Mantripragada and Holmans20 The evidence for six loci is based only on single, albeit very large, studies: deletions at 17q12, Reference Moreno-De-Luca, Mulle, Kaminsky, Sanders, Myers and Adam19 distal 16p11.2 Reference Guha, Rees, Darvasi, Ivanov, Ikeda and Bergen18 and 17p12 Reference Kirov, Grozeva, Norton, Ivanov, Mantripragada and Holmans20 and duplications at 1q21.1, Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 AS/PWS region Reference Ingason, Kirov, Giegling, Hansen, Isles and Jakobsen6 and at 16p13.11. Reference Ingason, Rujescu, Cichon, Sigurdsson, Sigmundsson and Pietilainen7 Moreover, the role of two loci, deletions at 15q11.2 and duplications at 16p13.11, has recently been challenged by a study that indicated that the rates among controls might be higher than originally reported, Reference Grozeva, Conrad, Barnes, Hurles, Owen and O'Donovan5 and no excess in individuals with schizophrenia was seen for these two CNVs in one of the largest studies. Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 Finally, each previous report focused on the identification of one, or only a small number of CNV loci, and several reports used partially overlapping data-sets. Consequently, the rate of all previously reported risk CNVs has not yet been evaluated in a single independent data-set that is not biased by the inclusion of the original discovery sample. We set out to evaluate these specific CNVs in a sample of 6882 people with schizophrenia that passed quality control. This represents the largest single CNV data-set yet reported in schizophrenia, and nearly doubles the sample size of patients from the total world literature for many loci. For control individuals, we used publicly available data from 6316 samples genotyped on arrays with a resolution similar to the case group. These data-sets are completely independent from samples used in previous studies or reviews that established the rates of these CNVs in schizophrenia.
Method
Samples
Case group
We collected data on patients with schizophrenia (case group) in two waves, which we call (a) CLOZUK and (b) CardiffCOGS (Cardiff Cognition in Schizophrenia). The CLOZUK sample (n = 6558) consists of individuals taking the antipsychotic clozapine. In the UK, clozapine is reserved for patients with treatment-resistant schizophrenia, who provide regular blood samples to allow detection of adverse drug effects. Through collaboration with Novartis, the manufacturer of a proprietary form of clozapine, we acquired anonymised blood samples from people on clozapine. Participants (71% male) were aged 18-90, with a recorded diagnosis of treatment-resistant schizophrenia according to the clozapine registration forms completed by their psychiatrists. The use of these anonymised samples for genetic association studies was approved by the local ethics committee. The CLOZUK sample has been described elsewhere. Reference Hamshere, Walters, Smith, Richards, Green and Grozeva21 The CardiffCOGS (n = 571) is a sample of patients with clinically diagnosed schizophrenia recruited from community, in-patient and voluntary sector mental health services in the UK. Interview with the Schedules for Clinical Assessment in Neuropsychiatry (SCAN) instrument Reference Wing, Babor, Brugha, Burke, Cooper and Giel22 and case-note review was used to arrive at a best-estimate lifetime diagnosis according to DSM-IV criteria. 23
Control group
As controls we used publicly available data downloaded from dbGAP (www.ncbi.nlm.nih.gov/gap). To avoid CNV detection biases between arrays with different probe densities and platforms, we chose data-sets genotyped with Illumina arrays that had a high overlap with the probes used to genotype the case group. The following data-sets were used: 1491 participants from the USA that took part in a study on smoking cessation, 3102 participants from the USA who took part in a study on melanoma and 1869 participants from Germany who took part in a study of refractive error (KORA study). Participants in the ‘smoking’ and ‘melanoma’ data-sets are cases or controls from these studies, whereas the KORA data-set is a population-based study where participants had refractive error measurements. The ethnicities of participants were derived from principal components analysis (PCA). A total of 91.4% of samples that passed quality control were from individuals of European ancestry. Full details on these data-sets are presented in the online supplement (section 1).
Genotyping and quality control filtering
Raw intensity files from each data-set were independently processed to account for potential batch effects. PennCNV Reference Wang, Li, Hadley, Liu, Glessner and Grant24 was used for CNV detection. To avoid cross-platform biases, we restricted CNV calling to the 520 766 probes present on all arrays used. Samples were excluded if any one of the following standard quality control statistics constituted an outlier within their source data-set: log R ratio (LRR) standard deviation, B-allele frequency (BAF) drift, wave factor (WF) and total number of CNVs (online supplement, section 2). Out of 13 591 samples with array data, 393 were excluded due to poor quality control or for being duplicates of the same individual after testing for identity-by-descent. The 8.6% non-European participants were retained in the analysis, to ensure that our data were comparable with those in recent reviews of CNV loci in schizophrenia. Reference Malhotra and Sebat14 The numbers and ethnicities of these participants are listed in online Table DS2. The final numbers after exclusions for quality control and duplicates was 6882 in the case group and 6316 in the control group. The quality control process for individual CNVs is detailed in the online supplement (section 2). Briefly, CNVs were included if they were >10 kb in size and had a frequency <1%, (applying filters with PLINK version 1.07). Reference Purcell, Neale, Todd-Brown, Thomas, Ferreira and Bender25 All CNVs were subsequently required to pass a median z-score outlier method of validation Reference Kirov, Pocklington, Holmans, Ivanov, Ikeda and Ruderfer26 that helps to remove false-positive CNVs, and to identify any missed CNVs.
Statistical analysis
For the analysis of CNV loci we used Fisher’s exact test (1-tailed as we were testing prior hypotheses). We performed a meta-analysis by adding our new data to those in the literature, applying a 2-tailed Fisher’s exact test to the combined data. As these CNVs have been shown to be subjected to strong negative selection pressure, their frequencies in the population essentially reflect the mutation rate and selection pressure operating against them. Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein1,Reference Rees, Moskvina, Owen, O'Donovan and Kirov27 They are therefore less likely to be subjected to population stratification caused by genetic drift, in the way common variants are (although there are some examples of ethnic differences in the mutation rates). Therefore, in the interests of clarity, we do not stratify the sample by ethnicity but provide the full breakdown of the data in the different ethnic groups in online Table DS3. The results do not change appreciably if they are restricted to only the European populations that comprise 91.4% of the sample.
There is no accepted convention as to what constitutes genome-wide significance for a CNV locus. Girirajan and colleagues Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein1 reported 72 recurrent CNVs that can cause a neurodevelopmental disorder, although in total, 120 genomic regions are potentially prone to recurrent CNVs because they are flanked by segments of high homology, called segmental duplications. Reference Girirajan, Dennis Megan, Baker, Malig, Coe and Campbell28 This suggests that a Bonferroni correction for multiple testing of recurrent CNVs (those flanked by segmental duplications) might require a P-value of <4.1×10–4 to be accepted as a significant association for this type of CNV (P = 0.05/120). Regarding associations with individual genes, a conservative Bonferroni correction would require correcting for the testing of ∼20 000 genes, or P<2.5×10–6 (P = 0.05/20 000).
Choice of CNVs for analysis
The list of previously implicated CNVs was taken from the largest meta-analysis to date. Reference Malhotra and Sebat14 To this list we added three loci: exon-disrupting deletions at the NRXN1 gene, as there is consensus that they increase risk for developing schizophrenia; Reference Kirov, Rujescu, Ingason, Collier, O'Donovan and Owen9,Reference Levinson, Duan, Oh, Wang, Sanders and Shi10,Reference Rujescu, Ingason, Cichon, Pietiläinen, Barnes and Toulopoulou13 deletions at distal 16p11.2, the evidence for which was published after the above review; Reference Guha, Rees, Darvasi, Ivanov, Ikeda and Bergen18 and duplications at the WBS region, a locus that just failed to reach significance in that review, but received support in a subsequent study. Reference Mulle, Pulver, McGrath, Wolyniec, Dodd and Cutler16 For duplications at the AS/PWS region, we tested their parental origin using a DNA methylation-sensitive high-resolution melting curve analysis, Reference Urraca, Davis, Cook, Schanen and Reiter29 as previous research suggested that the maternal ones are specifically implicated Reference Ingason, Kirov, Giegling, Hansen, Isles and Jakobsen6 (online supplement, section 5).
Results
The rates of CNVs among the case and control groups are presented in Table 1. For 13 out of the 15 CNVs, we found higher rates in the case than in the control group. For six of these, the difference was nominally significant in this new sample alone (Table 1).
Of the loci where previous evidence was modest, the most striking result was for AS/PWS duplications, where we found eight in patients and none in controls (P = 0.0055). When combined with previous data (Table 2, for a more detailed version that includes results from previous studies see online Table DS6), this CNV is now a strongly supported schizophrenia risk variant. Moreover, as this is an imprinted locus, we tested the parental origin of our eight duplications, and all were shown to be maternal in origin, similar to the original publication. Reference Ingason, Kirov, Giegling, Hansen, Isles and Jakobsen6 Duplication at 16p13.11, another previously weakly associated CNV, now also shows strong evidence in the combined data (Table 2).
Table 1 Findings from our data-set for previously implicated copy number variation (CNV) loci in schizophreniaFootnote a

| Case group (n = 6882) | Control group (n = 6316) | ||||||
|---|---|---|---|---|---|---|---|
| Locus | Position in Mb | CNVs, n | Frequency, % | CNVs, n | Frequency, % | OR (95% CI) | P |
| 1q21.1 del | chr1:146,57-147,39 | 12 | 0.17 | 1 | 0.016 | 11.03 (1.43-84.86) | 0.0027 |
| 1q21.1 dup | chr1:146,57-147,39 | 8 | 0.12 | 5 | 0.079 | 1.47 (0.48-4.49) | 0.35 |
| NRXN1 del | chr2:50,15-51,26 | 11 | 0.16 | 0 | 0.00 | NA (1.25-∞) | 7.7 × 10−4 |
| 3q29 del | chr3:195,73-197,34 | 4 | 0.058 | 0 | 0.00 | NA (0.44-∞) | 0.074 |
| WBS dup | chr7:72,74-74,14 | 3 | 0.044 | 1 | 0.016 | 2.75 (0.29-26.48) | 0.35 |
| VIPR2 dup | chr7:158,82-158,94 | 1 | 0.015 | 6 | 0.095 | 0.15 (0.02-1.27) | 0.99 |
| 15q11.2 del | chr15:22,80-23,09 | 44 | 0.64 | 26 | 0.41 | 1.56 (0.96-2.53) | 0.046 |
| AS/PWS dup | chr15:24,82-28,43 | 8 | 0.12 | 0 | 0.00 | NA (0.90-∞) | 0.0055 |
| 15q13.3 del | chr15:31,13-32,48 | 4 | 0.058 | 2 | 0.032 | 1.84 (0.34-10.03) | 0.38 |
| 16p13.11 dup | chr16:15,51-16,30 | 24 | 0.35 | 12 | 0.19 | 1.84 (0.92-3.68) | 0.056 |
| 16p11.2 distal del | chr16:28,82-29,05 | 0 | 0.00 | 2 | 0.032 | NA (0-3.82) | 1 |
| 16p11.2 dup | chr16:29,64-30,20 | 27 | 0.39 | 0 | 0.00 | NA (3.09-∞) | 2.3 ×10−8 |
| 17p12 del | chr17:14,16-15,43 | 4 | 0.058 | 3 | 0.047 | 1.22 (0.27-5.47) | 0.55 |
| 17q12 del | chr17:34,81-36,20 | 1 | 0.015 | 0 | 0.00 | NA (0.11-∞) | 0.52 |
| 22q11.2 del | chr22:19,02-20,26 | 20 | 0.29 | 0 | 0.00 | NA (2.28-∞) | 2.2 ×10−6 |
| Totals | 171 | 2.48 | 58 | 0.92 | 1.4 ×10−12 | ||
del, deletion; dup, duplications, NA, not applicable; WBS, Williams-Beuren syndrome; AS/PWS, Angelman/Prader-Willi syndrome.
a. Copy number variation positions are in UCSC Build 37. Significant results are in bold (using Fisher exact test, 1-tailed).
The only instances in our study where CNVs were more common in the control than in the case group concern duplications at VIPR2 and deletions at distal 16p11.2. In neither case is the excess of CNVs in the control group significant. In the case of VIPR2, meta-analysis is no longer supportive, whereas the evidence for association at distal 16p11.2 remains nominally significant (Table 2 and Table DS6). Analysis of only individuals of European descent gave essentially the same results. The distribution of CNVs in the different data-sets and ethnic groups is presented in Table DS3.
In the present sample, which is not subject to the potential bias of including the original studies that discovered the associations, about 2.5% of the case group and 0.9% of the control group carry one of the CNVs in Table 1, a highly significant excess (P = 1.4×10–12). Only four individuals in the case group carry two of these CNVs (online supplement, section 6).
Table 2 Combined results of previous studies and the current data-setFootnote a
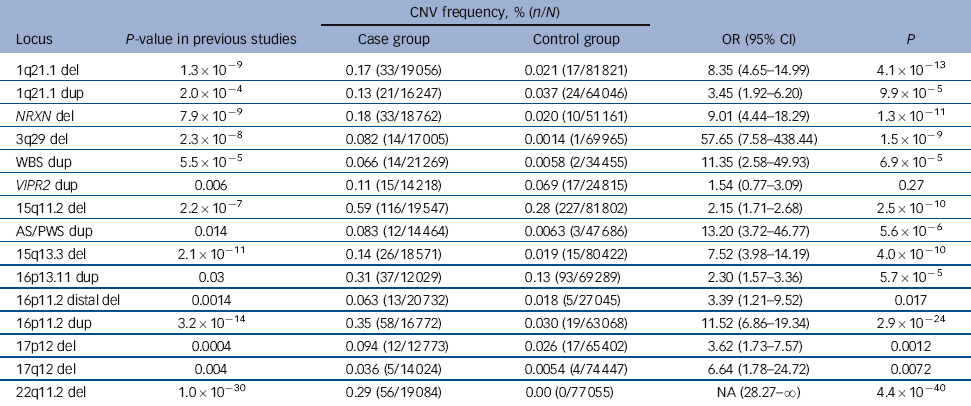
| CNV frequency, % (n/N) | |||||
|---|---|---|---|---|---|
| Locus | P-value in previous studies | Case group | Control group | OR (95% CI) | P |
| 1q21.1 del | 1.3×10−9 | 0.17 (33/19 056) | 0.021 (17/81 821) | 8.35 (4.65-14.99) | 4.1×10−13 |
| 1q21.1 dup | 2.0×10−4 | 0.13 (21/16 247) | 0.037 (24/64 046) | 3.45 (1.92-6.20) | 9.9×10−5 |
| NRXN del | 7.9×10−9 | 0.18 (33/18 762) | 0.020 (10/51 161) | 9.01 (4.44-18.29) | 1.3×10−11 |
| 3q29 del | 2.3×10−8 | 0.082 (14/17 005) | 0.0014 (1/69 965) | 57.65 (7.58-438.44) | 1.5×10−9 |
| WBS dup | 5.5×10−5 | 0.066 (14/21 269) | 0.0058 (2/34 455) | 11.35 (2.58-49.93) | 6.9×10−5 |
| VIPR2 dup | 0.006 | 0.11 (15/14 218) | 0.069 (17/24 815) | 1.54 (0.77-3.09) | 0.27 |
| 15q11.2 del | 2.2×10−7 | 0.59 (116/19 547) | 0.28 (227/81 802) | 2.15 (1.71-2.68) | 2.5×10−10 |
| AS/PWS dup | 0.014 | 0.083 (12/14 464) | 0.0063 (3/47 686) | 13.20 (3.72-46.77) | 5.6×10−6 |
| 15q13.3 del | 2.1×10−11 | 0.14 (26/18 571) | 0.019 (15/80 422) | 7.52 (3.98-14.19) | 4.0×10−10 |
| 16p13.11 dup | 0.03 | 0.31 (37/12 029) | 0.13 (93/69 289) | 2.30 (1.57-3.36) | 5.7×10−5 |
| 16p11.2 distal del | 0.0014 | 0.063 (13/20 732) | 0.018 (5/27 045) | 3.39 (1.21-9.52) | 0.017 |
| 16p11.2 dup | 3.2×10−14 | 0.35 (58/16 772) | 0.030 (19/63 068) | 11.52 (6.86-19.34) | 2.9×10−24 |
| 17p12 del | 0.0004 | 0.094 (12/12 773) | 0.026 (17/65 402) | 3.62 (1.73-7.57) | 0.0012 |
| 17q12 del | 0.004 | 0.036 (5/14 024) | 0.0054 (4/74 447) | 6.64 (1.78-24.72) | 0.0072 |
| 22q11.2 del | 1.0×10−30 | 0.29 (56/19 084) | 0.00 (0/77 055) | NA (28.27-∞) | 4.4×10−40 |
del, deletion; dup, duplications; NA, not applicable; WBS, Williams-Beuren syndrome; AS/PWS, Angelman/Prader-Willi syndrome.
a. For a more detailed version of this table that includes the CNV frequency, % (n/N) from previous studies see online Table DS6. P-values are based on Fisher exact test, 2-tailed.
In the analysis of all 15 loci in the combined data (Table 2), all but one of the CNVs showed significant evidence of association. For 11 of these, the significance surpasses the threshold for multiple testing correction that we suggest in the Method (P<4.1×10–4). For many of them the statistical significance is greatly improved compared with the previous results, most strikingly for 15q11.2, AS/PWS, 16p13.11, 16p11.2 and 22q11.2, where the P-values were strengthened by several orders of magnitude.
Discussion
In an analysis of the largest single schizophrenia sample to date, we establish more accurate estimates of risk from individual CNVs in an independent sample, and estimate the total burden of susceptibility conferred by this group of CNVs. The vast majority of patients in this study were recruited on the basis that they have a diagnosis of treatment-resistant schizophrenia according to their psychiatrist and were taking clozapine for that indication. The availability of clinician diagnoses allowed us to exclude the limited number of samples from individuals with diagnoses other than schizophrenia. The convention for psychiatric samples has been that patient inclusion is based on research diagnoses arrived at following detailed interview and phenotyping procedures (as is the case in the CardiffCOGS sample in this study). As genome analysis has become more affordable than the establishment of a formal research diagnosis, the latter has now become the limiting step for exploiting the microarray technology. The CLOZUK sampling method offers a pragmatic approach to recruit unusually large numbers of patients with schizophrenia, and nearly doubles the number of patients with schizophrenia analysed in the previous literature. The use of such samples is supported by evidence that with the use of operationalised criteria, clinician diagnoses of schizophrenia have high specificity and positive predictive values when validated against research-based approaches. Reference Ekholm, Ekholm, Adolfsson, Vares, Ösby and Sedvall30,Reference Jakobsen, Frederiksen, Hansen, Jansson, Parnas and Werge31 Furthermore, we have reported findings that support the validity of the individuals in CLOZUK as a schizophrenia sample with genetic data, by demonstrating that of the most strongly associated schizophrenia alleles in the Psychiatric Genetics Consortium Stage 1, 85% (66/78) showed the same direction of effect in the CLOZUK sample, sign test P = 1.7×10–10. Reference Hamshere, Walters, Smith, Richards, Green and Grozeva21 In the present study, the findings of very similar rates of susceptibility CNVs in the CLOZUK sample compared with previous samples (Table DS6), recruited using conventional methods, support the comparability of the two types of samples.
In order to reduce the potential bias of using different arrays, we used only Illumina platforms and only analysed those probes common to all arrays. We used the z-score method to both validate each CNV and check whether any CNV in the regions in Table 1 had been missed.
The current study provides support for most previously implicated CNVs, as we found higher rates in the case group than in the control group for 13 of the 15 loci. The support is particularly strong for duplications at 16p11.2 (P = 2.3×10–8) and at the AS/PWS critical region (P = 0.0055) and for deletions at 22q11.2 (P = 2.2×10–6), 1q21.1 (P = 0.0027), NRXN1 (P = 7.7×10–4) and 15q11.2 (P = 0.046). All eight duplications at the AS/PWS region were of maternal origin (online supplement, section 5), thus supporting the original report. Reference Ingason, Kirov, Giegling, Hansen, Isles and Jakobsen6 Two loci: deletions at 15q11.2 and duplications at 16p13.11, that were not supported in two recent papers, Reference Grozeva, Conrad, Barnes, Hurles, Owen and O'Donovan5,Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 also receive support in the current study and the statistical significance of their overall association with schizophrenia is strengthened by several orders of magnitude (Table 2).
Four of the loci in Table 2 do not surpass a significance threshold that corrects for the multiple testing of large recurrent CNVs (P<4.2×10–4), or for individual genes (P<2.5×10–6) (see Method): duplications at VIPR2, and deletions at distal 16p11.2, 17p12 and 17q12.
Burden of schizophrenia-associated CNVs
Overall, 2.5% of the case group v. 0.9% of the control group carry one or more of these CNVs. This is highly significant in this completely independent data-set (P = 1.4×10–12). They are associated with a range of odds ratios and each locus clearly makes a different contribution to the increase in risk (Table 2). They are also known to increase risk for other disorders, such as epilepsy (15q11.2 Reference de, Trucks, Helbig, Mefford, Baker and Leu32 and 15q13.3 Reference Dibbens, Mullen, Helbig, Mefford, Bayly and Bellows33 ), congenital heart disease (1q21.1 Reference Christiansen, Dyck, Elyas, Lilley, Bamforth and Hicks34 and 22q11.2 Reference Botto, May, Fernhoff, Correa, Coleman and Rasmussen35 ), attention-deficit hyperactivity disorder (16p13.11 Reference Williams, Zaharieva, Martin, Langley, Mantripragada and Fossdal36 ) and obesity (distal 16p11.2 Reference Bochukova, Huang, Keogh, Henning, Purmann and Blaszczyk37 ), and all but two (at VIPR2 and 17p12) increase risk for developmental delay and autism spectrum disorders. Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein1 The overall contribution is modest but the effect size is sufficiently large to suggest that if seen in a patient, it is very likely to be relevant to the disorder, although not sufficient to account for the disease.
Summary of the findings for the individual loci
1q21.1 deletions and duplications
Deletions at 1q21.1 were among the first implicated loci. 8,Reference Stefansson, Rujescu, Cichon, Pietilainen, Ingason and Steinberg38 Our new data provide strong support for their role in schizophrenia (P = 0.0027), with a frequency in the case group identical to that in previous reports (0.17%), Reference Malhotra and Sebat14 confirming an approximately eightfold excess of deletions among patients, with an extremely strong statistical support of P = 4.1×10–13 in the combined literature (Table 2). Duplications at this locus have only been implicated in one study, Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 but with the addition of our data (although not significant on its own), the evidence for duplications at this locus in schizophrenia is now stronger (P = 9.9×10–5).
NRXN1 deletions
The gene NRXN1 encodes for a presynaptic cell adhesion protein, which binds with postsynaptic proteins called neuroligins and plays a vital role in the formation, maintenance and release of neurotransmitters in synapses. Reference Südhof39 Exonic deletions disrupting this gene have been consistently implicated in schizophrenia Reference Kirov, Rujescu, Ingason, Collier, O'Donovan and Owen9,Reference Levinson, Duan, Oh, Wang, Sanders and Shi10,Reference Rujescu, Ingason, Cichon, Pietiläinen, Barnes and Toulopoulou13,Reference Kirov, Gumus, Chen, Norton, Georgieva and Sari40 and autism spectrum disorder. Reference Glessner, Wang, Cai, Korvatska, Kim and Wood41 The current study confirms their role, as we found 11 exonic deletions in the case group (0.16%) and none in the control group, P = 7.7×10–4. This brings the overall significance in the combined literature to P = 1.3×10–11 (easily surpassing our multiple testing correction threshold for individual genes of P<2.5×10–6), with approximately a ninefold excess in the case group (Table 2). The positions of exon-disrupting CNVs at this locus in our new data-set are shown in the online supplement, section 4.
3q29 deletions
The role of this CNV in schizophrenia was first reported by Mulle et al, Reference Mulle, Dodd, McGrath, Wolyniec, Mitchell and Shetty12 and confirmed by Levinson et al. Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 The finding of four individuals in the case group with deletions and none in the control group just fails to reach significance in our independent sample (P = 0.074) but can be regarded as supportive independent confirmation. With an overall significance of P = 1.5×10–9, it is another locus where the evidence for a role in schizophrenia is very strong. Only one such deletion has been found in nearly 70 000 controls, indicating it is highly penetrant (Table 2).
WBS duplications
The reciprocal duplication of the WBS region was first implicated as increasing the risk for autism. Reference Sanders, Ercan-Sencicek, Hus, Luo, Murtha and Moreno-De-Luca42 The region was implicated in schizophrenia after the finding of a de novo duplication Reference Kirov, Pocklington, Holmans, Ivanov, Ikeda and Ruderfer26 and reached statistical support in a large collaborative study. Reference Mulle, Pulver, McGrath, Wolyniec, Dodd and Cutler16 The finding of three individuals in the case group and one in the control group with this duplication in our study constitutes only modest support, but the overall strength of the evidence remains strong, at P = 6.9×10–5.
VIPR2 duplications
The evidence for duplications disrupting this gene comes from two studies that used largely overlapping samples. Reference Levinson, Duan, Oh, Wang, Sanders and Shi10,Reference Vacic, McCarthy, Malhotra, Murray, Chou and Peoples17 The overall evidence from the previous literature was modest: P = 0.006. Reference Malhotra and Sebat14 As we found duplications in six people in the control group and only one in the case group, the evidence in favour of this locus is no longer significant in the combined literature (P = 0.27). The positions of CNVs at this locus in our new data-set are shown in the online supplement (section 4). Most CNVs at this gene are large and covered with a high number of probes on the arrays (medians of 381 kb and 66 probes, details in online Table DS4), therefore they should be called reliably on these arrays. We also examined the region with all available probes for CNV calling on the different arrays, and with the z-score method, and found no additional CNVs that had been missed. The rate in our control group is slightly higher than in previous studies (0.095% v. 0.059%, Table 1 and Table DS6), although this difference is not significant (P = 0.4).
15q11.2 deletions
This was one of the first CNVs implicated in schizophrenia. Reference Kirov, Grozeva, Norton, Ivanov, Mantripragada and Holmans20,Reference Stefansson, Rujescu, Cichon, Pietilainen, Ingason and Steinberg38 However, a recent report of a higher rate in controls than that reported in the original paper Reference Grozeva, Conrad, Barnes, Hurles, Owen and O'Donovan5 and the lack of support in the study by Levinson et al Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 clearly indicated the need for replication. Here we found independent significant evidence for association (P = 0.046), that strengthens the evidence in the combined analysis to P = 2.5×10–10 (Table 2).
AS/PWS duplications
The modest prior evidence for the role of this duplication of the AS/PWS critical region in schizophrenia comes from a single publication with just four observations in patients. Reference Ingason, Kirov, Giegling, Hansen, Isles and Jakobsen6 We found another eight CNVs in our case group and none in the control group (P = 0.0055), thus substantially strengthening this finding. Even more notably, by using DNA methylation-sensitive high-resolution melting-curve analysis of the SNRPN locus, Reference Urraca, Davis, Cook, Schanen and Reiter29 which does not require DNA from the parents, we found that all eight duplications are of maternal origin, as in the original study. These findings further underline the importance of imprinted genes (those genes subject to parent of origin specific epigenetic regulation) in the aetiology of psychosis and other neurodevelopmental disorders. Reference Wilkinson, Davies and Isles43 These duplications are among the most common genetic susceptibility factors for autism spectrum disorder, where they are found in nearly 1:500 cases. Reference Moreno-De-Luca, Sanders, Willsey, Mulle, Lowe and Geschwind44
15q13.3 deletion
This is also among the first implicated CNVs in schizophrenia, 8,Reference Stefansson, Rujescu, Cichon, Pietilainen, Ingason and Steinberg38 and received further strong support in the study by Levinson et al. Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 It was found at a ∼tenfold higher rate in patients with schizophrenia, with a strong statistical support in the previous literature, Reference Malhotra and Sebat14 P = 2.1×10–11. Although we found only a twofold excess in our case group in our new sample, at 0.058% v. 0.032%, P = 0.38, its role as a susceptibility factor for schizophrenia remains very strong in the combined literature (P = 4.0×10–10). This locus also increases the risk for epilepsy. Reference Dibbens, Mullen, Helbig, Mefford, Bayly and Bellows33,Reference Helbig, Hartmann and Mefford45
16p13.11 duplication
The previous evidence for this CNV comes mostly from a single study Reference Ingason, Rujescu, Cichon, Sigurdsson, Sigmundsson and Pietilainen7 and the overall number of analysed patients with schizophrenia so far, at 5147 individuals, is smaller than in our new data-set. Two recent studies Reference Grozeva, Conrad, Barnes, Hurles, Owen and O'Donovan5,Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 weakened the evidence for this locus, and in the recent review by Malhotra & Sebat Reference Malhotra and Sebat14 it had very weak statistical support, P = 0.03. Here we found 24 CNVs in the case group and 12 in the control group, an excess that just fails to reach significance, P = 0.056. Combined with the earlier data, our study strengthens the statistical support for this locus in schizophrenia to P = 5.7×10–5. This CNV has also been implicated in attention-deficit hyperactivity disorder. Reference Williams, Zaharieva, Martin, Langley, Mantripragada and Fossdal36
16p11.2 duplication
This is our strongest finding, with a P = 2.3×10–8 in the discovery sample alone, and a combined evidence at P = 2.9×10–24, with an odds ratio of over 11. This duplication is also one of the strongest autism spectrum disorder CNV risk factors. Reference Moreno-De-Luca, Sanders, Willsey, Mulle, Lowe and Geschwind44
16p11.2 distal deletion
This CNV was suggested to confer susceptibility to schizophrenia by Guha et al. Reference Guha, Rees, Darvasi, Ivanov, Ikeda and Bergen18 We found no support for this locus in the present study, with two deletions in the control group and none in the case group. However, the lack of deletions among our case group could be as a result of ascertainment bias. Obesity is found in at least 50% of 16p11.2 distal deletion carriers. Reference Bachmann-Gagescu, Mefford, Cowan, Glew, Hing and Wallace46 Clozapine produces the most severe weight gain among all antipsychotics Reference Leadbetter, Shutty, Pavalonis, Vieweg, Higgins and Downs47 and the most common cause for the reluctance of UK psychiatrists to prescribe clozapine is the potential weight gain. Reference Nielsen, Dahm, Lublin and Taylor48 It is therefore possible that psychiatrists are less likely to give clozapine to carriers of 16p11.2 distal deletion (as such patients are more likely to be obese already), thus potentially reducing the frequency of this CNV in the CLOZUK sample. The combined result from the literature retains the statistical support for the role of this locus (P = 0.017), but it clearly requires testing in further data-sets.
17p12 deletion
This deletion causes the neurological disorder hereditary neuropathy with liability to pressure palsies and was implicated in schizophrenia on the basis of only eight observations in the original case group. Reference Kirov, Grozeva, Norton, Ivanov, Mantripragada and Holmans20 Although the overall statistical evidence is still in favour of it increasing the risk for schizophrenia (P = 0.0012), the support is not compelling, raising the need for further replication. It does not increase the risk for either developmental delay or autism, Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein1 unlike most other loci discussed here, and it is possible that the original report was a false-positive finding.
17q12 deletion
Originally this locus had been known to cause renal cysts and diabetes. It was identified as a susceptibility CNV for schizophrenia with only four observations in the case group Reference Moreno-De-Luca, Mulle, Kaminsky, Sanders, Myers and Adam19 in a study that also implicated its role in autism spectrum disorder. We found only one deletion in a patient, a rate a quarter of that in the original study. Reference Moreno-De-Luca, Mulle, Kaminsky, Sanders, Myers and Adam19 Although we found no deletions in the control group, the overall P-value of all data is not sufficiently robust to definitively conclude that this is a schizophrenia susceptibility locus (P = 0.0072). Therefore, this locus also requires testing in further schizophrenia data-sets.
22q11.2 deletion
This was the first CNV implicated in schizophrenia Reference Karayiorgou, Morris, Morrow, Shprintzen, Goldberg and Borrow2,Reference Shprintzen, Goldberg, Golding-Kushner and Marion49 and has received extensive replication over the years. Reference Levinson, Duan, Oh, Wang, Sanders and Shi10 It affects ∼40 genes and leads to a variety of physical anomalies. Reference Shprintzen50 The latest review found a rate of 0.30% in patients and 0% in controls. Reference Malhotra and Sebat14 This rate is practically identical to our new sample, where we found 20 carriers in the case group (0.29%) and none in the control group. It remains the most significantly associated CNV in schizophrenia: P = 4.4×10–40.
Implications
Out of 15 previously implicated CNV loci, 11 are now strongly associated with schizophrenia from the combined results of the previous literature and our new data. The evidence for the remaining four loci should be regarded as still equivocal and requiring further investigation. Our findings indicate that approximately 2.5% of individuals with schizophrenia carry at least one known pathogenic CNV. The odds ratios of these CNVs in relation to schizophrenia range between ∼2 and >50 and nearly all of them are also associated with a range of other neurodevelopmental disorders, such as autism spectrum disorder and intellectual deficit. Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein1 Moreover, a number of the individual pathogenic CNVs are associated with particular physical disease phenotypes such as epilepsy (15q11.2 and 15q13.3), congenital heart disease (1q21.1 and 22q11.2), microcephaly (1q21.1, 3q29 and 16p11.2) and obesity (16p11.2 distal). Reference Girirajan, Rosenfeld, Coe, Parikh, Friedman and Goldstein1,Reference de, Trucks, Helbig, Mefford, Baker and Leu32-Reference Bochukova, Huang, Keogh, Henning, Purmann and Blaszczyk37,Reference Helbig, Hartmann and Mefford45,Reference Mefford, Sharp, Baker, Itsara, Jiang and Buysse51 Given their frequency, these findings therefore suggest that routine screening for CNVs should be made available and that the results will have immediate implications for genetic counselling, and given their comorbidity with other medical disorders, for patient management as well. The robust identification of 11 relatively high penetrance risk alleles for schizophrenia also offers promise for biological research aimed at developing animal and cellular models for the identification of novel disease mechanisms and drug targets.
Acknowledgements
We thank the participants and clinicians who took part in the CardiffCOGS study. We acknowledge Andrew Iles, David Parslow, Carissa Philipart and Sophie Canton for their work in recruitment, interviewing and rating. For the CLOZUK sample we thank Novartis for their guidance and cooperation. We also thank staff at The Doctor’s Laboratory, in particular Lisa Levett and Andrew Levett, for help and advice regarding sample acquisition. We acknowledge Kiran Mantripragada, Lesley Bates, Catherine Bresner and Lucinda Hopkins for laboratory sample management.
The authors acknowledge the contribution of data from outside sources: (a) Genetic Architecture of Smoking and Smoking Cessation accessed through dbGAP: Study Accession: phs000404.v1.p1. Funding support for genotyping, which was performed at the Center for Inherited Disease Research (CIDR), was provided by 1 X01 HG005274-01 (CIDR is fully funded through a federal contract from the National Institutes of Health to The Johns Hopkins University, contract number HHSN268200782096C). Assistance with genotype cleaning, as well as with general study coordination, was provided by the Gene Environment Association Studies (GENEVA) Coordinating Center (U01 HG004446). Funding support for collection of data-sets and samples was provided by the Collaborative Genetic Study of Nicotine Dependence (COGEND; P01 CA089392) and the University of Wisconsin Transdisciplinary Tobacco Use Research Center (P50 DA019706, P50 CA084724). (b) High Density SNP Association Analysis of Melanoma: Case-Control and Outcomes Investigation, dbGaP Study Accession: phs000187.v1.p1: research support to collect data and develop an application to support this project was provided by 3P50CA093459, 5P50CA097007, 5R01ES011740, and 5R01CA133996. (c) Genetic Epidemiology of Refractive Error in the KORA (Kooperative Gesundheitsforschung in der Region Augsburg) Study, dbGaP Study Accession: phs000303.v1.p1. Principal investigators: Dwight Stambolian, University of Pennsylvania, Philadelphia, Pennyslavian, USA; H. Erich Wichmann, Institut für Humangenetik, Helmholtz-Zentrum München, Germany; National Eye Institute, National Institutes of Health, Bethesda, Maryland, USA. Funded by R01 EY020483, National Institutes of Health, Bethesda, Maryland, USA.




eLetters
No eLetters have been published for this article.