


The most robust risk factor predicting bipolar disorder is a positive family history. Reference Gottesman, Laursen, Bertelsen and Mortensen1 This familial clustering has a strong genetic basis. Reference Farmer, Elkin and McGuffin2,Reference Hamshere, O'Donovan, Jones, Jones, Kirov and Green3 Adolescence appears to mark the beginning of the high-risk period for the onset of major mood episodes associated with bipolar disorder. Reference Duffy4-Reference Angst and Sellaro6 However, there may be earlier non-specific antecedents Reference Shaw, Egeland, Endicott, Allen and Hostetter7 and heterogeneity of the disorder may influence the illness trajectory. Reference McGuffin, Perroud, Uher, Butler, Aitchison and Craig8,Reference Alda9 Long-term response to lithium has been used to identify a more homogeneous subtype of bipolar disorder with characteristic clinical, familial, treatment and neurobiological findings. Reference Grof10,Reference Grof, Duffy, Alda and Hajek11
Over the past 20 years, there has been growing interest in longitudinal studies of the children of parents with bipolar disorder, recognising the importance of mapping the early clinical course over development. Although earlier cross-sectional high-risk studies reported an increased lifetime risk of a confusing breadth of psychopathology in offspring of affected parents, Reference Duffy, Doucette, Lewitzka, Alda, Hajek and Grof12,Reference DelBello and Geller13 longitudinal prospective studies have provided convergent evidence that bipolar disorder-related mood episodes typically debut as depressive episodes in adolescence. Reference Shaw, Egeland, Endicott, Allen and Hostetter7,Reference Hillegers, Reichart, Wals, Verhulst, Ormel and Nolen14,Reference Duffy, Alda, Hajek and Grof15 Mania has remained a rare observation in prospectively followed high-risk children, only appearing later in adolescence and early adulthood. Furthermore, these longitudinal studies have clarified that the high rates of reported attention-deficit hyperactivity disorder (ADHD) in some offspring studies likely reflect the general psychopathological burden of the family and in most cases are not an antecedent to typical lithium-responsive bipolar disorder. Reference Duffy16
In this paper we provide a comprehensive analysis of the evolution of psychopathology at the syndrome level from an ongoing prospective study of the offspring of parents with bipolar disorder and test a clinical staging model previously proposed. This study has been conducted over 17 years and earlier analyses were carried out and published. Since our last publication on this topic, Reference Duffy, Alda, Hajek, Sherry and Grof17 we have observed this cohort for 3 additional years and added more eligible offspring from within identified pedigrees as they became available (dynamic cohort). Specifically, with a larger number of high-risk offspring being observed over a longer period we: (a) estimated the differential risk of lifetime psychopathology between high-risk and control offspring; (b) compared the clinical course of mood disorders between high-risk subgroups (defined by the lithium response of the parent); and (c) tested the evidence of a clinical staging model using adjusted multistate models.
Method
Identification of families
Detailed methods of this study have been published elsewhere. Reference Duffy, Alda, Crawford, Milin and Grof18 Briefly, affected parents were identified through their involvement in neurobiological research based in Ottawa, Ontario and Halifax, Nova Scotia, Canada. High-risk offspring were eligible if they had one parent with a confirmed DSM-IV 19 diagnosis of bipolar disorder type I on the basis of Schedule for Affective Disorders and Schizophrenia - lifetime version (SADS-L) Reference Endicott and Spitzer20 interviews conducted by a research psychiatrist. In 23% of cases (53/229), we expanded recruitment within the families to include a first-degree relative of the original bipolar disorder proband, who themselves had a diagnosis of either bipolar disorder type I or type II or recurrent major depressive disorder. In all cases, the other parent was confirmed to have no lifetime history of major psychiatric disorder at enrolment (major mood, psychosis, substance use disorder) based on SADS-L interview.
Control offspring were recruited from schools in Ottawa. Families with children in grades 6-12 were mailed a demographic screening questionnaire by the school. Parents from interested families who mailed back these questionnaires were invited to a clinical assessment following the SADS-L format conducted by a research psychiatrist. Control offspring were eligible if both of their parents were confirmed to have no lifetime history of major psychiatric disorder at the time of enrolment. All clinical diagnoses in parents were reviewed and confirmed on the basis of masked consensus involving two additional research psychiatrists.
Offspring
As part of this ongoing prospective study, high-risk and control offspring between the ages of 7 and 25 years were assessed in accordance with research protocol. All offspring were assessed by a psychiatrist masked to family affiliation at baseline and subsequently annually (on average) or anytime symptoms developed using the Kiddie Schedule for Affective Disorders and Schizophrenia - present and lifetime version (KSADS-PL)/SADS-L format interviews (depending on their age). DSM-IV diagnoses were based on best estimate procedures using all available clinical information and reviewed on a consensus basis by two additional research psychiatrists masked to family affiliation.
Statistical approach
To test for differences between groups, we used two-sided t-tests for continuous outcomes and Fisher’s exact tests for categorical outcomes. Hazard ratios (HRs) were estimated using Cox proportional hazard models, with the exact method for handling ties. These models account for censoring and variable age of last assessment. When zero members of a group had a diagnosis, hazard ratios were estimated using the method of Firth, with the Breslow method for handling ties.
Lifetime prevalence was estimated using cumulative incidence functions, Reference Kalbfleisch and Prentice21 which account for censoring and variable age of last assessment. As the oldest member of the sample was still in early adulthood, cumulative incidence very likely underestimates lifetime prevalence until all offspring have passed through the period of risk. To adjust for sibling correlation, we used linear mixed models for continuous outcomes and generalised estimating equations (GEEs) for categorical outcomes.
In order to assess the validity of our previously proposed staging model, we fitted multistate models to the data using the msm package (version 1.1) in R. 22 Three different types of models were assessed: an unrestricted model that allowed individuals to transition between any stages (skipping, moving forward and backward); a forward model that only allowed individuals to transition into higher stages (moving forward but allowed skipping); and a progressive model that only allowed transitions from one stage to the next stage (moving forward without skipping). As some individuals were observed to experience the stages out of order, the models included estimated misclassifications. The different models were assessed using Akaike information criterion (AIC) scores. Reference Sakamoto, Ishiguro and Kitagawa23 In comparing two models, a lower AIC score indicated a better fit to the data. To test for a difference in evolution of staging in offspring of lithium responder v. lithium non-responder parents, a chi-squared test with simulation-based P-value was performed in R. Reference Team24
Table 1 Characteristics of high-risk and control offspring

| High-risk
offspring (n = 229) |
Control
offspring (n = 86) |
P | LiR offspring (n = 96) |
LiNR
offspring (n = 133) |
P | |
|---|---|---|---|---|---|---|
| Gender of offspring, % | ||||||
| Male | 39.74 | 41.86 | 0.797Footnote a | 36.46 | 42.11 | 0.414Footnote a |
| Female | 60.26 | 58.14 | 63.54 | 57.89 | ||
| Parent with bipolar disorder, % | ||||||
| Father | 48.03 | - | - | 42.71 | 51.88 | 0.182Footnote a |
| Mother | 51.96 | - | 57.29 | 48.12 | ||
| Hollingshead SES, % | ||||||
| 1 | 0.44 | 0 | 0.0171a* | 1.04 | 0 | <0.001a* |
| 2 | 2.62 | 0 | 0 | 4.51 | ||
| 3 | 7.86 | 3.49 | 12.50 | 4.51 | ||
| 4 | 38.43 | 25.58 | 28.13 | 45.86 | ||
| 5 | 50.66 | 69.77 | 58.33 | 45.11 | ||
| Age at first assessment, years: mean (s.d.) | 16.35 (5.34) | 14.71 (2.25) | 0.006Footnote b Footnote * | 16.79 (5.93) | 16.02 (4.87) | 0.278Footnote b |
| Age at last assessment, years: mean (s.d.) | 22.56 (6.82) | 19.10 (3.21) | <0.0001Footnote b Footnote * | 23.18 (7.53) | 22.15 (6.23) | 0.2614Footnote b |
| Follow-up duration, years: mean (range) | 6.29 (0-15.75) | 4.39 (0-7.93) | <0.0001Footnote b Footnote * | 6.38 (0-15.75) | 6.13 (0-15.14) | 0.6630Footnote b |
| GAF score at last assessment, mean (s.d.) | 81.56 (11.15) | 86.79 (8.18) | <0.0001Footnote b Footnote * | 84.71 (9.55) | 74.82 (11.90) | <0.0001Footnote b Footnote * |
LiR, offspring of lithium responder parent; LiNR, offspring of lithium non-responder parent; SES, socioeconomic status; GAF, Global Assessment of Functioning.
a. Fisher’s exact test.
b. t-test.
* P<0.05.
All analysis was performed using the SAS software version 9.3 for Windows 7 unless otherwise specified, and were adjusted for gender, age and socioeconomic status (SES) based on the Hollingshead Scale Reference Hollingshead25 and sibling correlation.
Results
Description of high-risk and control offspring
For this analysis, we included data from 229 high-risk offspring from 113 families and 86 control offspring from 55 families. As shown in Table 1, the high-risk families were typically from middle to upper SES intact families, with parents educated to at least the postgraduate level. The control families were more tightly clustered in the higher SES strata compared with the high-risk families. The distribution was more weighted towards the higher SES strata in the lithium responder compared with lithium non-responder families. Comparison offspring were on average somewhat younger than high-risk offspring.
Lifetime psychopathology among high-risk and control offspring
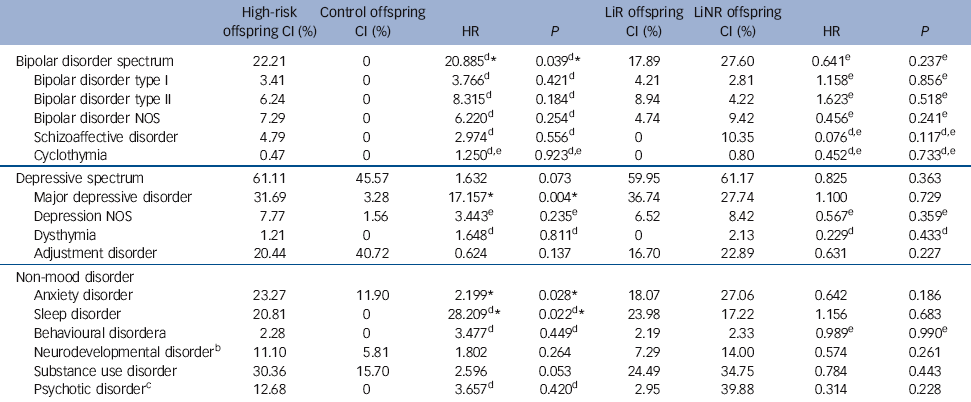
The hazard of major mood disorders in the high-risk compared with the control offspring was elevated several fold (Table 2). Specifically, the estimated HR of bipolar spectrum disorders and of major depressive disorder was 20.89 (P = 0.04) and 17.16 (P = 0.004) respectively in high-risk offspring compared with controls. In the high-risk offspring, the median age at onset for bipolar spectrum disorders was concentrated in late adolescence and early adulthood, whereas the median age of depressive spectrum disorders ranged from mid to late adolescence (Table 3). There were no significant differences in the risk of diagnosable mood disorders between high-risk subgroups (lithium responder v. lithium non-responder), with the notable exception that all cases of schizoaffective disorder occurred among the offspring of lithium non-responder parents. Control offspring had relatively high rates of minor mood disorders, largely attributable to the diagnosis of adjustment disorder (symptoms associated with a specific undesirable life event).
Table 2 Cumulative incidence (CI) of lifetime DSM-IV diagnoses
| High-risk
offspring CI (%) |
Control
offspring CI (%) |
HR | P | LiR offspring CI (%) |
LiNR
offspring CI (%) |
HR | P | |
|---|---|---|---|---|---|---|---|---|
| Bipolar disorder spectrum | 22.21 | 0 | 20.885Footnote d Footnote * | 0.039Footnote d Footnote * | 17.89 | 27.60 | 0.641Footnote e | 0.237Footnote e |
| Bipolar disorder type I | 3.41 | 0 | 3.766Footnote d | 0.421Footnote d | 4.21 | 2.81 | 1.158Footnote e | 0.856Footnote e |
| Bipolar disorder type II | 6.24 | 0 | 8.315Footnote d | 0.184Footnote d | 8.94 | 4.22 | 1.623Footnote e | 0.518Footnote e |
| Bipolar disorder NOS | 7.29 | 0 | 6.220Footnote d | 0.254Footnote d | 4.74 | 9.42 | 0.456Footnote e | 0.241Footnote e |
| Schizoaffective disorder | 4.79 | 0 | 2.974Footnote d | 0.556Footnote d | 0 | 10.35 | 0.076Footnote d , Footnote e | 0.117Footnote d , Footnote e |
| Cyclothymia | 0.47 | 0 | 1.250Footnote d , Footnote e | 0.923Footnote d , Footnote e | 0 | 0.80 | 0.452Footnote d , Footnote e | 0.733Footnote d , Footnote e |
| Depressive spectrum | 61.11 | 45.57 | 1.632 | 0.073 | 59.95 | 61.17 | 0.825 | 0.363 |
| Major depressive disorder | 31.69 | 3.28 | 17.157Footnote * | 0.004Footnote * | 36.74 | 27.74 | 1.100 | 0.729 |
| Depression NOS | 7.77 | 1.56 | 3.443Footnote e | 0.235Footnote e | 6.52 | 8.42 | 0.567Footnote e | 0.359Footnote e |
| Dysthymia | 1.21 | 0 | 1.648Footnote d | 0.811Footnote d | 0 | 2.13 | 0.229Footnote d | 0.433Footnote d |
| Adjustment disorder | 20.44 | 40.72 | 0.624 | 0.137 | 16.70 | 22.89 | 0.631 | 0.227 |
| Non-mood disorder | ||||||||
| Anxiety disorder | 23.27 | 11.90 | 2.199Footnote * | 0.028Footnote * | 18.07 | 27.06 | 0.642 | 0.186 |
| Sleep disorder | 20.81 | 0 | 28.209Footnote d Footnote * | 0.022Footnote d Footnote * | 23.98 | 17.22 | 1.156 | 0.683 |
| Behavioural disorderFootnote a | 2.28 | 0 | 3.477Footnote d | 0.449Footnote d | 2.19 | 2.33 | 0.989Footnote e | 0.990Footnote e |
| Neurodevelopmental disorderFootnote b | 11.10 | 5.81 | 1.802 | 0.264 | 7.29 | 14.00 | 0.574 | 0.261 |
| Substance use disorder | 30.36 | 15.70 | 2.596 | 0.053 | 24.49 | 34.75 | 0.784 | 0.443 |
| Psychotic disorderFootnote c | 12.68 | 0 | 3.657Footnote d | 0.420Footnote d | 2.95 | 39.88 | 0.314 | 0.228 |
HR, hazard ratio; LiR, offspring of lithium responder parent; LiNR, offspring of lithium non-responder parent; NOS, not otherwise specified.
a. Behavioural disorders include oppositional defiant disorder and conduct disorder.
b. Neurodevelopmental disorders include attention-deficit hyperactivity disorder, learning disorder and Cluster A traits.
c. Psychotic disorders include schizophrenia, psychosis NOS, schizoid and schizotypal disorder.
d. Firth’s method with Breslow’s method for handling ties.
e. Not adjusted for socioeconomic status.
* P<0.05.
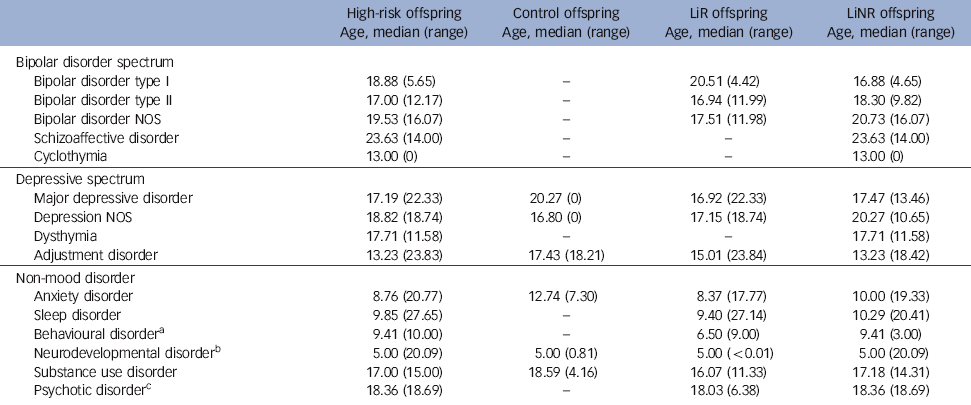
Table 3 Median age at onset of DSM-IV diagnoses
| High-risk
offspring Age, median (range) |
Control
offspring Age, median (range) |
LiR offspring Age, median (range) |
LiNR
offspring Age, median (range) |
|
|---|---|---|---|---|
| Bipolar disorder spectrum | ||||
| Bipolar disorder type I | 18.88 (5.65) | - | 20.51 (4.42) | 16.88 (4.65) |
| Bipolar disorder type II | 17.00 (12.17) | - | 16.94 (11.99) | 18.30 (9.82) |
| Bipolar disorder NOS | 19.53 (16.07) | - | 17.51 (11.98) | 20.73 (16.07) |
| Schizoaffective disorder | 23.63 (14.00) | - | - | 23.63 (14.00) |
| Cyclothymia | 13.00 (0) | - | - | 13.00 (0) |
| Depressive spectrum | ||||
| Major depressive disorder | 17.19 (22.33) | 20.27 (0) | 16.92 (22.33) | 17.47 (13.46) |
| Depression NOS | 18.82 (18.74) | 16.80 (0) | 17.15 (18.74) | 20.27 (10.65) |
| Dysthymia | 17.71 (11.58) | - | - | 17.71 (11.58) |
| Adjustment disorder | 13.23 (23.83) | 17.43 (18.21) | 15.01 (23.84) | 13.23 (18.42) |
| Non-mood disorder | ||||
| Anxiety disorder | 8.76 (20.77) | 12.74 (7.30) | 8.37 (17.77) | 10.00 (19.33) |
| Sleep disorder | 9.85 (27.65) | - | 9.40 (27.14) | 10.29 (20.41) |
| Behavioural disorderFootnote a | 9.41 (10.00) | - | 6.50 (9.00) | 9.41 (3.00) |
| Neurodevelopmental disorderFootnote b | 5.00 (20.09) | 5.00 (0.81) | 5.00 (<0.01) | 5.00 (20.09) |
| Substance use disorder | 17.00 (15.00) | 18.59 (4.16) | 16.07 (11.33) | 17.18 (14.31) |
| Psychotic disorderFootnote c | 18.36 (18.69) | - | 18.03 (6.38) | 18.36 (18.69) |
LiR, offspring of lithium responder parent; LiNR, offspring of lithium non-responder parent; NOS, depression not otherwise specified.
a. Behavioural disorders include oppositional defiant disorder and conduct disorder.
b. Neurodevelopmental disorders include attention-deficit hyperactivity disorder, learning disorder and Cluster A traits.
c. Psychotic spectrum disorders include schizophrenia, psychosis NOS, schizoid and schizotypal disorder.
When estimating the risk of any DSM-IV non-mood disorder, the high-risk offspring had a higher lifetime risk of anxiety (HR = 2.20, P = 0.03) and sleep disorders (HR = 28.21, P = 0.02), with marginal evidence of an increased risk of substance use disorders (HR = 2.60, P = 0.05) compared with control offspring (Table 2). Interestingly, anxiety disorders tended to occur significantly earlier in the high-risk compared with the control offspring (mean 9.78 years (s.d. = 4.23) v. 13.85 years (s.d.= 2.58), P = 0.02). Also, there was an increase in the lifetime risk of neurodevelopmental disorders (ADHD, learning disabilities, Cluster A traits) in high-risk compared with control offspring, with double the rate in the lithium non-responder compared with lithium responder subgroup (14% v. 7.29%); however, this did not reach statistical significance.
An exploratory analysis found no evidence that having a family history of anxiety disorders affected the risk of the high-risk offspring developing anxiety disorders (HR = 0.84, P = 0.74). However, there was evidence that having a family history of substance use disorders increased the risk of developing a substance use disorder in the high-risk offspring (HR = 2.01, P = 0.01).
Early course of mood disorders in the high-risk offspring
The high-risk subgroups differed significantly in the nature of the clinical course of diagnosable mood disorders; namely, the affected offspring of lithium responder parents were more likely to have an episodic clinical course with complete clinical remissions between mood episodes, than were the offspring of lithium non-responder parents (GEE odds ratio (OR) 8.37; P = 0.01). However, there was no evidence of a difference in the lifetime risk of major (HR = 1.10, P = 0.69) or minor mood disorders (HR = 0.74, P = 0.23), nor was there evidence of a difference in the age at onset of index mood disorders (β = 0.78, P = 0.45) between the high-risk subgroups. Similarly, there was no difference between high-risk subgroups in the hazard of life-time psychotic features (HR = 0.49, P = 0.13) or in the odds of being admitted to hospital (OR = 0.53, P = 0.36). However, there was evidence of lower Global Assessment of Functioning at last observation among the offspring of lithium non-responder parents compared with the offspring of lithium responder parents (β = 5.94, P = 0.001).
Comparison between parent and child course of major mood disorder
In exploratory analyses there was no evidence that the parents’ age at onset of index mood episode had an effect on the offspring’s age at onset of index mood episode (β = 0.06, P = 0.32) and no evidence that the polarity of the parent’s index mood episode affected the polarity of the offspring’s index mood episode (OR = 3.34, P = 0.24). Similarly, there was no evidence that a parent’s lifetime psychotic features predicted the offspring’s lifetime psychotic features (OR = 2.10, P = 0.12).
Early course of bipolar disorder in high-risk offspring
Of the 31/229 high-risk offspring (13.54%) who met DSM-IV lifetime diagnostic criteria for a bipolar spectrum disorder (bipolar disorder types I and II, bipolar disorder not otherwise specified (NOS), schizoaffective disorder), the majority had an index diagnosable mood episode in the depressive polarity (26/31, 83.87%). Furthermore, in those whose mood disorder began with a depressive episode, the first diagnosable activated episode did not occur until 20.85 years of age (s.d. = 4.82) on average. Consistent with previous reports, there was no case of diagnosable mania or hypomania observed prior to age 15.5 years, and the earliest age at which an offspring met the DSM diagnosis for bipolar disorder NOS was 12.5 years. The mean age at onset of index mood episode in the offspring of lithium non-responder parents was somewhat lower (15.19 years, s.d. = 4.24) compared with offspring of lithium responder parents (17.19 years, s.d. = 3.83), although this was not statistically significant (P = 0.188).
Clinical stages in the development of bipolar disorder in high-risk offspring
Based on our observations in our last publication, we speculated that bipolar disorder might evolve in a series of reliable stages starting with non-specific non-mood disorders in childhood (i.e. anxiety), followed by minor mood and adjustment disorders in early adolescence, then major depressive episodes and finally hypomanic/manic episodes. Reference Duffy, Alda, Hajek, Sherry and Grof17 Therefore, here we used adjusted survival analysis to determine the risk of developing a major mood episode (major depressive disorder, hypomania or mania) in those high-risk offspring compared with those without a preceding non-mood diagnosis. From this analysis there was evidence that a childhood anxiety disorder significantly increased the hazard of developing a major mood episode (HR = 1.84, P = 0.02). However, neither childhood sleep disorders nor neurodevelopmental disorders were associated with a statistically significantly increased risk of developing a major mood episode (HR = 1.00, P = 0.10 and HR = 1.35, P = 0.46 respectively).
From the AIC scores of the multistate models using a narrow definition of stage 4 (bipolar disorder types I and II or schizoaffective bipolar disorder) there was evidence that the progressive model was sufficient when compared with the other two more complex models (unrestricted AIC: 2299; forward AIC: 2247; progressive AIC: 2094; see Fig. 1). The data are consistent with the transition through the stages in order without skipping. There was no evidence that stage transitions differed between the lithium responder and lithium non-responder subgroups (χ2 = 7.98, P = 0.45).
In an exploratory analysis including bipolar disorder NOS as part of the stage 4 definition, there was evidence that the progressive model was sufficient when compared with the other two more complex models (unrestricted AIC: 2518; forward AIC: 2279; progressive AIC: 2145). Comparing the different definitions of stage 4, the first definition (without bipolar disorder NOS) appeared to fit the data better, producing multistate models with lower AIC scores.
Discussion
In this study we describe the latest observations pertaining to the natural course of bipolar disorder in an ongoing prospective study of the offspring of well-characterised parents with bipolar disorder. The first aim was to estimate and compare the lifetime risk of diagnosable disorders between the high-risk and control offspring and between the two high-risk subgroups defined by the affected parent’s long-term response to lithium (response v. no response). The adjusted cumulative incidence of bipolar disorder and depressive spectrum disorders was 22.2% and 61.1% respectively in the high-risk offspring. This represents a substantial increase from our last published observations, Reference Duffy, Alda, Hajek, Sherry and Grof17 while adjusting here for important confounders not previously considered. In this analysis, there were no striking differences in lifetime risk of mood disorders between the high-risk subgroups, with the exception that all five cases of schizoaffective disorder occurred in offspring from parents who did not respond to lithium prophylaxis. This suggests that offspring of lithium non-responder parents may have a vulnerability to develop psychotic spectrum as well as mood disorders. Furthermore, with longer observation time, we reported an increased risk of diagnosable depressive spectrum disorders among control offspring. However, these were largely depressive and anxiety symptoms associated with an identifiable stressor (i.e. adjustment disorder) and not a major depressive disorder as seen in the high-risk offspring.
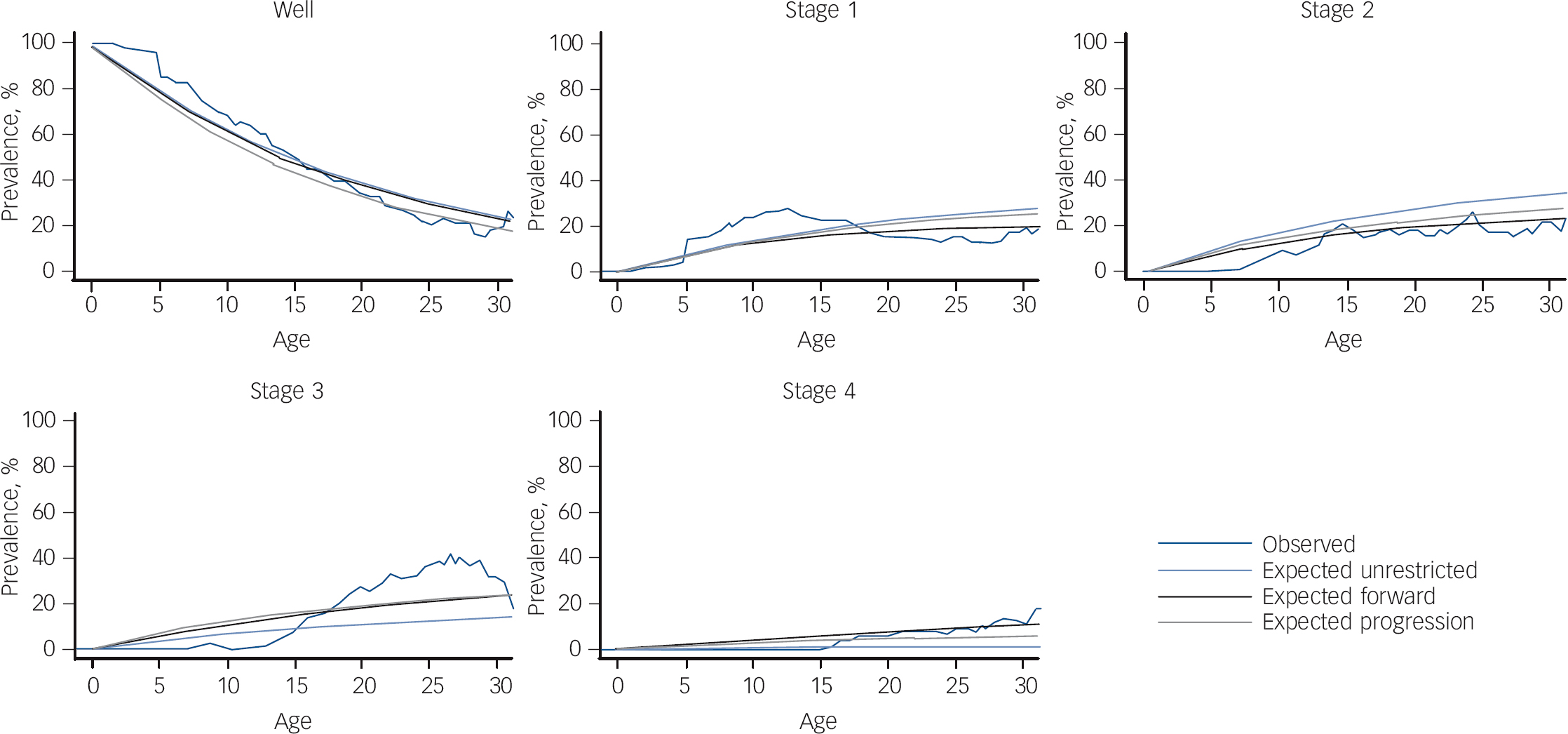
Fig. 1 Multistate models testing progression through clinical stages.
In terms of non-mood lifetime psychopathology, we found an elevated risk of childhood anxiety and sleep disorders among high-risk compared with control offspring; the latter of which fell short of statistical significance in in earlier analyses. Interestingly, the mean age at onset of anxiety disorders was significantly earlier in the high-risk compared with control offspring. This is consistent with the speculation of a more vulnerable central nervous system. Consistent with prior publications by ourselves and others, Reference Duffy, Horrocks, Milin, Doucette, Persson and Grof26 there was an increased risk of substance use disorders among the high-risk offspring (HR = 2.596, P = 0.053) at a mean age of 17.46 years (s.d. = 3.12); convergent with the timing of the onset of mood disorders.
The second aim was to describe the early course of diagnosable mood disorders in the high-risk offspring, and to compare between the high-risk subgroups. To date, 13 offspring of lithium responder and 18 offspring of lithium non-responder parents have met lifetime criteria for diagnosable bipolar disorder. Consistent with previous reports, in the majority of cases the index mood episode was depressive, occurring in mid adolescence (16.03 years, s.d. = 4.12); whereas the index activated episode, clinically declaring the bipolar diathesis, typically occurred several years after the first depressive episode at a mean age of 20.02 years (s.d. = 4.98). Consistent with other longitudinal high-risk studies, Reference Shaw, Egeland, Endicott, Allen and Hostetter7,Reference Hillegers, Reichart, Wals, Verhulst, Ormel and Nolen14 we did not observe diagnosable cases of hypomania or mania during childhood. This lack of observed childhood mania occurred in the context of confirmed familial risk with multiple affected members in successive generations. This suggests that typically the manic polarity of bipolar disorder does not manifest until at least adolescence.
After adjusting for age, gender, SES and familial clustering, there was evidence of a predictive relationship between anxiety disorders in childhood and the subsequent development of major mood disorders in the high-risk offspring. This observation is consistent with an earlier unadjusted estimation. Reference Duffy, Alda, Hajek, Sherry and Grof17 Importantly, in this analysis we presented new evidence that childhood anxiety disorders in this high-risk offspring cohort were not attributable to an increased risk of anxiety disorders in adult family members. This finding supports the speculation that clinically significant anxiety disorders are related to the bipolar disorder diathesis in these high-risk children. This speculation is further supported from recent reports of antecedent anxiety in other studies of offspring of parents with bipolar disorder. Reference Nurnberger, McInnis, Reich, Kastelic, Wilcox and Glowinski27
In terms of early predictors of the bipolar disorder diathesis in high-risk children, it is likely that sleep (i.e. circadian disorders), and in certain subgroups neurodevelopmental disorders, also confer an increased risk of bipolar disorder spectrum psychopathology; however, we have not yet the power to show a statistically significant association. In a different paper, the relationship between ADHD and bipolar disorder in the lithium non-responder cohort is more fully discussed. Reference Duffy16 Essentially, evidence from this study and from other longitudinal studies suggest that neurodevelopmental disorders may be antecedents to psychotic spectrum illnesses, including atypical or psychotic spectrum bipolar disorder and that this may have a specific genetic basis. Reference St Clair28
Our third aim was to test the ‘staging hypothesis’ that we proposed based on our earlier observations and assessed initially using a simple sign test. Reference Duffy, Alda, Hajek and Grof15,Reference Duffy, Alda, Hajek, Sherry and Grof17 With longer observation and using more modelling techniques, we found evidence that high-risk offspring who go on to develop bipolar disorder do so in a predictable forward clinical sequence consistent with clinical staging. Not all offspring manifest every stage; however, once they enter the model, they do so in a forward sequence and tend not to skip stages (see Fig. 2). The offspring of lithium non-responder parents tended to differ from the offspring of lithium responder parents in the breadth of stage 1 and stage 4 psychopathology and in the nature of the clinical course of mood disorders (lithium responder parents have episodic mood disorders and lithium non-responder parents have non-episodic/non-fully remitting disorders). Specifically, the offspring of lithium non-responder parents manifest neurodevelopmental disorders, along with sleep and anxiety disorders in stage 1 and psychotic as well as bipolar disorders in stage 4. The latter observation reflects the longer observation period allowing for more offspring to transition to end-stage illness. Given the increased risk of psychotic disorders in the extended family members of lithium non-responder bipolar disorder probands, Reference Grof10,Reference Grof, Alda, Grof, Zvolsky and Walsh29 this observation is consistent with a the hypothesis of aetiological heterogeneity of bipolar disorder; that is, a different underlying genetic contribution to lithium responder compared with lithium non-responder subtypes. Reference Grof, Alda, Grof, Zvolsky and Walsh29

Fig. 2 Clinical staging model of bipolar disorder in high-risk offspring subgroups.
Limitations of the study
There are several limitations in regard to the analyses presented here that should be taken into consideration. First, the findings are in a very good agreement with several studies that used comparable methodology; however, they differ from those investigations that used alternative approaches to recruitment, assessment and diagnosis (see Duffy et al Reference Duffy, Doucette, Lewitzka, Alda, Hajek and Grof12 for review). Second, eligible offspring are enrolled at variable ages and are followed for variable lengths of time. Therefore, there is an element of retrospective data collection that varies depending on the age at which individuals enter the study. However, recall bias related to clinical information is minimised by considering all available clinical information (observations made in real time and later reviewed) and by interviewing both the offspring and their parents at relatively frequent intervals. In the same way, there is potential recall bias related to parent clinical data, which may have limited the strength of the associations between parent and offspring clinical course. Although psychiatrists were masked to family affiliation when first assessing the offspring, masking was more difficult to maintain over subsequent assessments. However, potential bias is minimised given that final diagnoses are made on a masked consensus review basis. In keeping with genetic studies, we expanded within pedigrees to include affected first-degree relatives (with bipolar disorder or recurrent major depressive disorder) of the original bipolar disorder type I probands as parents in this study. Additionally, although this is a naturalistic study, some offspring are clinically treated at least for a period of time. We estimated that less than 20% of acute mood episodes were treated and less than 20% of intervals between episodes were exposed to mood-stabilising medication. Reference Duffy, Alda, Hajek and Grof15 Therefore, medication in this cohort is thought to have a minimal effect on the early clinical course. Finally, the staging model is based on full-threshold diagnoses and we have not as yet analysed the effect of adding clinically significant symptoms (i.e. manic or depressed) to the model.
Clinical implications
The findings presented here have important implications for advancing both clinical and research efforts directed at improving early accurate diagnosis of bipolar disorder and identifying important early intervention targets; especially when viewed in light of complementary findings from other independent longitudinal high-risk studies. First, as in other areas of medicine, we need to adopt a developmental approach to diagnosis, asking about the early natural history and evolution of psychopathology in individual patients. Second, we should be aware that bipolar disorder very often occurs in the context of familial risk and therefore a detailed family history should be taken and used to determine the possible trajectory of illness in individual symptomatic adolescent patients and their treatment response profile. That is, family history provides an important lens or perspective through which to view otherwise non-specific clinical presentations. Third, as initially described by McGorry and colleagues in the context of schizophrenia, Reference McGorry, Nelson, Goldstone and Yung30 a clinical staging model provides an important framework to guide early intervention and prevention efforts as well as research investigations. As in other areas of medicine, a clinical staging framework presents a distinct advance over cross-sectional symptom-based approaches to diagnosis.
Lastly, these findings raise a provocative question as to the nature of the underlying pathophysiological processes and specifically why the surface clinical manifestations are relatively non-specific early in development. Translational research efforts targeting changes in biomarkers across the developmental course and clinical stages of illness within well-characterised samples of high-risk youth may provide important insights into these questions.
Acknowledgements
This work would not be possible without the continued commitment of our incredibly motivated and inspiring research families.






eLetters
No eLetters have been published for this article.