


Although mania is the defining feature of bipolar I disorder, depressive episodes pose the major burden of this condition. Patients with bipolar I disorder experience syndromal or subsyndromal depressive symptoms three times more commonly than syndromal/subsyndromal manic symptoms. Reference Hlastala, Frank, Mallinger, Thase, Ritenour and Kupfer1–Reference Kupka, Altshuler, Nolen, Suppes, Luckenbaugh and Leverich3 Depressive symptoms in bipolar I disorder are associated with poor functioning Reference Kauer-Sant'Anna, Bond, Lam and Yatham4 and poorer quality of life Reference Yatham, Lecrubier, Fieve, Davis, Harris and Krishnan5 and increased risk of suicide. Reference Angst, Hengartner, Gamma and Angst6,Reference Osby, Brandt, Correia, Ekbom and Sparen7 Yet, only three treatments (quetiapine; olanzapine plus fluoxetine combination; lurasidone) have been approved by the US Food and Drug Administration for treatment of acute bipolar depression. In real-world clinical practice, a significant proportion of patients either does not respond to or does not tolerate the side-effects of these treatments. Thus, other treatments that are safe and effective are urgently needed. Antidepressants in combination with mood stabilisers or atypical antipsychotics are widely used for treating bipolar depression in clinical practice. Reference Baldessarini, Leahy, Arcona, Gause, Zhang and Hennen8,Reference Vieta, Langosch, Figueira, Souery, Blasco-Colmenares and Medina9 Yet, the efficacy of such strategy continues to be controversial. Reference Pacchiarotti, Bond, Baldessarini, Nolen, Grunze and Licht10 Three large double-blind trials examined the efficacy of antidepressant add-on v. placebo add-on to mood stabilisers in treating bipolar depression. Fluoxetine plus olanzapine was more effective than not only placebo alone but also placebo plus olanzapine in improving bipolar depressive symptoms. Reference Tohen, Vieta, Calabrese, Ketter, Sachs and Bowden11 Paroxetine was no more effective than placebo when added to lithium, but superiority of paroxetine to placebo was noted in a post hoc analysis of patients with lower serum lithium levels. Reference Nemeroff, Evans, Gyulai, Sachs, Bowden and Gergel12 In the STEP-BD study, durable recovery rates were no different between the paroxetine or bupropion plus mood stabiliser group compared with the placebo plus mood stabiliser group. Reference Sachs, Nierenberg, Calabrese, Marangell, Wisniewski and Gyulai13 Since this study did not have a placebo monotherapy arm, the results could be interpreted to indicate that both treatments were either equally effective or ineffective in improving depressive symptoms. If it was indeed a failed study, it highlights a recognised problem with patient selection for placebo-controlled studies in mood disorders, which is the recruitment of an assay sensitive population. Reference Vieta and Cruz14 High placebo response rates and high drop-out rates greatly reduce assay sensitivity. There is now mounting evidence that conduct of trials at non-academic sites may further compromise the quality of recruitment. Reference Merlo-Pich, Alexander, Fava and Gomeni15,Reference Dunlop, Thase, Wun, Fayyad, Guico-Pabia and Musgnung16
Overall there is a paucity of studies comparing the addition of antidepressant v. placebo to a mood stabiliser or an atypical antipsychotic as a strategy for managing bipolar depression. Reference Vieta, Locklear, Gunther, Ekman, Miltenburger and Chatterton17 Agomelatine represents a novel pharmacology among antidepressants as it is the only compound possessing both melatonin receptor (MT1 and MT2) agonist and 5-hydroxytryptamine 2 C (5-HT2c) receptor antagonist properties. Reference De, Guardiola-Lemaitre, Mocaer, Renard, Munoz and Millan18 The acute and long-term efficacy and tolerability of agomelatine has been clearly demonstrated at doses of 25–50 mg in patients with major depressive disorder Reference Hickie and Rogers19–Reference Goodwin, Boyer, Emsley, Rouillon and De21 and in those with generalised anxiety disorders. Reference Stein, Ahokas and De22,Reference Stein, Ahokas, Albarran, Olivier and Allgulander23 The efficacy and safety of agomelatine in patients with major depressive disorder and generalised anxiety disorders compared favourably with other antidepressants. Reference Stein, Ahokas, Albarran, Olivier and Allgulander23,Reference Kasper, Corruble, Hale, Lemoine, Montgomery and Quera-Salva24 In a preliminary open-label study that assessed the efficacy and safety of agomelatine 25 mg in addition to lithium or valproate in patients with bipolar I depression, a high rate of treatment response (81%) was obtained over a 6-week treatment period, together with good acceptability. Reference Calabrese, Guelfi and Perdrizet-Chevallier25 The primary objective of the present study was to further explore the antidepressant efficacy of 8 weeks of treatment with agomelatine 25–50 mg as an add-on to a mood stabiliser (MS) lithium or valproate (AGO-MS) compared with placebo add-on to a mood stabiliser (PBO-MS) in patients with bipolar I disorder in a current major depressive episode. The secondary objectives were (a) to assess the safety of AGO-MS and (b) to collect preliminary data on AGO-MS maintenance of efficacy and long-term safety over a 10-month double-blind extension treatment period.
Method
This was an international, double-blind, randomised study using a flexible dosage of agomelatine or placebo in combination with a mood stabliser (lithium or valproate), conducted in 67 centres in 15 countries (Argentina, Australia, Austria, Brazil, Canada, Denmark, Finland, France, Germany, India, Korea, Netherlands, Poland, South Africa, Spain) from July 2006 to December 2008 (trial registration: ISRCTN28588282). The study was performed in accordance with the principles of Good Clinical Practice E6 of the International Conference of Harmonisation (CPMP/ICH/135/95) and the Declaration of Helsinki, Finland. The study was approved by the relevant local ethics committees. All participants provided written informed consent for participation in the study.
Study population
Male or female out-patients eligible for this study were aged 18 (at least 21 years old in Argentina) to 70 years and presented with a bipolar I disorder currently in a major depressive episode according to DSM-IV-TR 26 criteria, confirmed with the Mini International Neuropsychiatric Interview. Reference Sheehan, Lecrubier, Sheehan, Amorim, Janavs and Weiller27 The current major depressive episode must have lasted at least 4 weeks but no more than 12 months. At the selection visit, they had to meet the following criteria: 17-item Hamilton Rating Scale for Depression (HRSD-17) Reference Hamilton28 total score ≥18 and Young Mania Rating Scale (YMRS) Reference Young, Biggs, Ziegler and Meyer29 total score 8.
Patients had to be on either lithium or valproic acid treatment for a minimum of 6 weeks before selection visit with a blood level of lithium between 0.5 and 1.0 mEq/L or valproic acid between 50 and 100 g/mL, tested between selection and inclusion visits. No mood stabiliser dose adjustment was allowed after inclusion, unless the serum levels were out of the therapeutic range. In order to ensure stable mood stabiliser concentrations within the accepted therapeutic range over the course of the study, mood stabiliser blood levels were measured between weeks 6 and 8, between months 5 and 6, and between months 10 and 12. Lorazepam use was allowed as needed between week 0 and week 2 visits only.
Patients were required to be physically healthy or to have stable medical illnesses on the basis of medical history, physical examination, 12-lead electrocardiogram (ECG) and clinical laboratory tests (biochemistry and haematology). Patients were excluded if they had transaminase values >3 times the upper normal limit, a blood creatinine value higher than 150 μmol/L, abnormal thyroid function and/or positive plasma beta-human chorionic gonadotropin.
Patients who met criteria for any of the following DSM-IV-TR disorders were excluded: obsessive–compulsive disorder, alcohol or drug misuse or dependence within the past 6 months. Patients were also excluded if they were at risk of suicide based on the assessment of the investigator or had a rating of four points on item three of HRSD-17. Patients were excluded if they had not responded to the administration of an appropriate dose of two different previous antidepressant treatments at the recommended dosage for the current episode. Patients with neurological disorders (dementia, seizure and stroke) or severe or uncontrolled organic disorders (neoplastic, cardiovascular, pulmonary or digestive disorders, unstable type 1 or 2 diabetes, hypo- or hyperthyroidism, untreated or uncontrolled hypertension) were also excluded. Patients were also excluded if they had received any of the following recent/concomitant therapies: formal psychotherapy, sleep deprivation and light therapy started within 2 weeks, electroconvulsive therapy (ECT) or transcranial magnetic stimulation for the current episode. For psychotropic treatments, a wash-out period of five elimination half-lives of the molecule was required before inclusion.
Study design and allocation to treatment
Patients had a 2- to 10-day run-in selection period between selection and inclusion (week 0) visits. At week 0, eligible patients were assigned to agomelatine or placebo according to a balanced (non-adaptive) randomisation with stratification based on the mood stabiliser (lithium or valproic acid) and the clinical centre. The treatment allocation and the dose increase were done centrally using an interactive voice response system (IVRS) in masked conditions for patients and investigators. From week 0, patients received agomelatine 25 mg/day or placebo. In case of insufficient improvement – determined using masked criteria defined by the sponsor prior to the study start – the dosage of agomelatine was increased to 50 mg/day after 2 weeks. Both the investigator and the patient were masked to dose adjustment. The 8-week double-blind treatment period was followed by an optional 10-month extension period. After study treatment discontinuation, regardless of when it occurred, there was a 1-week follow-up period without study treatment.
Throughout the duration of the study, patients took two tablets orally once a day in the evening (around 20.00 h). Patients received either agomelatine 25 mg (1 tablet of 25 mg + 1 placebo tablet), agomelatine 50 mg (2 tablets of 25 mg) or placebo (2 placebo tablets). The dosage schedule, the appearance of the study medication and the taste were the same from inclusion to the end of the treatment period for all patients. Tablets were packaged in identical blisters with identical labelling.
Efficacy measurements
In order to protect against baseline score inflation, the HRSD-17 was used to determine study eligibility, whereas change in the total Montgomery–Åsberg Depression Rating Scale (MADRS) Reference Montgomery and Asberg30 total score from baseline to the last post-baseline value over the acute 8-week treatment period was used as the primary efficacy measure. The MADRS was administered at each visit by the study investigators.
Secondary efficacy measures included response to treatment (defined as a decrease from baseline in MADRS total score 50%) over the 8-week and 12-month periods, changes in the HRSD-25 (HRSD-17 + 8 items of Reverse Vegetative Symptom Scale), the Clinical Global Impressions scale for Bipolar Disorder – Modified (CGI-BP-M), Reference Vieta Pascual, Torrent Font, Martínez-Arán, Colom Victoriano, Reinares Gabnepen and Benabarre Hernández31 the Hamilton Rating Scale for Anxiety (HRSA), Reference Hamilton32 the Leeds Sleep Evaluation Questionnaire (LSEQ) Reference Parrott and Hindmarch33 and the Quality of Life Enjoyment and Satisfaction Questionnaire – Short Form (QLESQ). Reference Endicott, Nee, Harrison and Blumenthal34
Safety measurements
Tolerability and safety evaluations were based on emergent adverse events spontaneously reported by the patient at each visit. Investigators were prompted to report any significant mood elevation according to their clinical judgement as an adverse event. Then, at least 1 week after the onset of the symptoms, the investigators were asked to describe the event by assessing the DSM-IV criteria for mania (hypomanic or manic) and to provide their final diagnosis. YMRS was rated by the investigator at selection, inclusion and each visit up to month 12.
The tolerability and safety evaluations were also based on 12-lead ECG abnormalities, biological samplings and physical examination. Vital signs (body weight, supine heart rate and blood pressure) were assessed at inclusion, week 8 and month 12 or at termination in the case of premature withdrawal.
Sample size calculations
Sample size calculations were performed based on the expected difference in MADRS change scores between baseline and last post-baseline visits over week 0 to week 8 between agomelatine and placebo groups in the full analysis set (using the two-sided Student's t-test for independent samples at 5% type I error). It was estimated that 148 patients per treatment group would provide a power of at least 80% if the difference in MADRS scores is at least 3 points with a standard deviation of 9 points. For a similar standard deviation, this number of patients is sufficient to conclude the superiority with a power of at least 90% if the difference is at least 3.5 points.
Statistical analysis
As established in the protocol registration, all efficacy analyses were performed on the full analysis set (intention-to-treat principle) based on the last post-baseline value recorded over the acute 8-week treatment period. The superiority of AGO-MS as compared with PBO-MS was examined on the MADRS total score using a three-way analysis of covariance with treatment and type of mood stabiliser as fixed effects, centre as random effect, with baseline MADRS total score as covariate and without interaction. A similar analysis was performed on the secondary outcome of HRSD-17 total score. In addition, an unadjusted analysis, based on the two-sided Student's t-test for independent samples, was performed.
The difference in response rates (as determined by 50% or greater reduction in MADRS scores was estimated, taking into account the last post-baseline value over the acute 8-week treatment period) between AGO-MS and PBO-MS was computed using a chi-squared test. For other comparisons, appropriate parametric and non-parametric statistics were used. Safety data were provided by treatment group over the acute and extension periods for the safety set of patients (i.e. having taken at least one dose of study treatment).
Post hoc analysis
In order to understand the impact of placebo response, we performed a post hoc analysis that included patients from all centres that had randomised at least one patient to each treatment condition (AGO-MS and PBO-MS). We then excluded all patient data from centres that had particular placebo or agomelatine responder rates (responder defined as ≥50 improvement on MADRS) i.e. 100%, ≥90%, ≥80%….0%. We examined changes in MADRS scores using a three-way analysis of covariance with treatment and type of mood stabiliser as a fixed effect, centre as a random effect and baseline MADRS total score as a covariate and without interaction. Statistical analysis was performed on SAS software, version 9.1. The type I error was set at 5% (two-sided tests).
Results
Clinical and demographic characteristics
Of the 405 patients selected, 344 patients (84.9%) were randomly allocated to receive AGO-MS (172 patients) or PBO-MS (172 patients) (Fig. 1). There were no significant differences in demographic characteristics between patients treated with AGO-MS or PBO-MS (Table 1). The average age of the randomised patients was 45.2 years (s.d. = 12.6) and 61.1% were female.

Fig. 1 Disposition of patients.
Table 1 Baseline characteristics for the randomised patients

| AGO-MS
group (n = 172) |
PBO-MS
group (n = 172) |
|
|---|---|---|
| Age, years: mean (s.d.) | 45.6 (13.3) | 44.7 (12.0) |
| Male/female, % | 39/61 | 39/61 |
| Bipolar I disorder | ||
| Duration, years: mean (s.d.) | 17.7 (11.3) | 15.8 (11.1) |
| Number of manic episodes, a mean (s.d.) | 2.5 (2.6) | 2.7 (2.7) |
| Number of mixed episodes, a mean (s.d.) | 0.6 (1.2) | 0.9 (2.0) |
| Major depressive episode | ||
| DSM-IV severity, % | ||
| Mild | 2.3 | 0.6 |
| Moderate | 59.3 | 55.8 |
| Severe without psychotic feature | 38.4 | 43.6 |
| Melancholic features, % | 64.5 | 75.0 |
| Current major depressive episode duration, months: mean (s.d.) | 3.2 (2.4) | 3.2 (2.3) |
| Number of previous depressive episodes, mean (s.d.) | 5.2 (5.6) | 4.6 (4.1) |
| Previous psychotropic treatments, b % | 83.7 | 82 |
| Lorazepam, % | 24.42 | 20.59 |
AGO-MS, agomelatine as an add-on to a mood stabiliser; PBO-MS, placebo as an add-on to a mood stabiliser.
a. Within the previous 10 years.
b. Within the last 12 months, other than lithium and valproic acid.
The average duration of bipolar I disorder in study participants was 16.7 years (s.d.= 11.2, median 14.6). The first episode was more frequently a depressive episode (62.5% of patients). The study patients had experienced 2.6 manic episodes (s.d. = 2.7), 0.8 mixed episodes (s.d. = 1.6) and 4.9 depressive episodes (s.d. = 4.9) during the past 10 years. During the past 12 months, the mean number of mood episodes was 1.7 (s.d. = 0.9). Throughout the study, mood stabiliser blood levels were within the range planned in the protocol (∼0.7 mmol/L for lithium; between 66 and 79 g/mL for valproic acid).
No clinically relevant differences between the groups were observed regarding physical examination, ECG and other baseline characteristics. Baseline scores for various measures including MADRS, HRSD, HRSA, YMRS, CGI-BP-M depression, mania or general score and QLESQ at inclusion are provided in Table 2. A total of 87 patients out of the 157 (55.4%) randomised to the AGO-MS group and continuing in the study after week 2 had a dose increase.
Table 2 Baseline rating scale scores for randomised patients

| Mean (s.d.) | ||
|---|---|---|
| AGO-MS
group (n = 172) |
PBO-MS
group (n = 172) |
|
| Montgomery–Åsberg Depression Rating Scale, total score | 29.5 (5.5) | 30.6 (5.2) |
| Hamilton Rating Scale for Depression 17-item total score | 24.9 (3.8) | 25.1 (3.7) |
| Hamilton Rating Scale for Depression – Reverse Vegetative Symptom Scale, total score (8-item) | 4.9 (3.0) | 5.0 (3.1) |
| Clinical Global Impressions scale for Bipolar Disorder – Modified | ||
| Depression score | 4.6 (0.8) | 4.7 (0.8) |
| Mania score | 1.1 (0.3) | 1.1 (0.3) |
| Bipolar disorder score a | 4.4 (0.9) | 4.5 (0.9) |
| Hamilton Rating Scale for Anxiety, total score | 20.2 (7.1) | 20.3 (6.3) |
| Young Mania Rating Scale, total score | 2.7 (1.7) | 2.9 (1.8) |
| Quality of Life Enjoyment and Satisfaction Questionnaire, general activities score | 37.1 (14.7) | 37.2 (14.4) |
AGO-MS, agomelatine as an add-on to a mood stabiliser; PBO-MS, placebo as an add-on to a mood stabiliser.
a. Longitudinal severity of bipolar disorder during the previous year.
Efficacy on depression
MADRS total score
Over the 8-week period, mean changes from baseline to last post-baseline value in the full analysis set were clinically relevant and identical in both groups (Table 3). Improvement on the MADRS total score was not significantly different between groups after adjustment for mood stabiliser, baseline score and centre (mean difference: −0.65 (s.e. = 1.13), P = −0.565). There was, correspondingly, no significant difference between the AGO-MS and PBO-MS groups in the proportion of responders (61.9% and 60.8%, respectively, P = 0.837) or remitters defined as a MADRS total score 12, (53.0% and 53.2% in the AGO-MS and PBO-MS groups, respectively).
Table 3 Mean Montgomery–Åsberg Depression Rating Scale (MADRS) scores and changes from baseline to last post-baseline value over the acute and extension treatment periods in the agomelatine as an add-on to a mood stabiliser (AGO-MS, n = 168) and placebo as an add-on to a mood stabiliser (PBO-MS, n = 171) groups (full analysis set)

| Mean (s.d.) | |||||
|---|---|---|---|---|---|
| MADRS total score | Baseline | Week 8 | Change over the
acute treatment period |
Month 12 | Change over the
extension treatment period |
| AGO-MS group | 29.5 (5.4) | 14.1 (10.2) | −15.4 (10.3) | 14.1 (12.8) | −15.4 (12.7) |
| PBO-MS group | 30.6 (5.2) | 15.4 (12.1) | −15.2 (11.8) | 15.7 (14.1) | −14.9 (13.6) |
A total of 244 patients (AGO-MS: 124; PBO-MS: 120) entered the 10-month extension phase and had at least one post week 8 MADRS score. There were no significant differences in MADRS change scores from baseline to end-point for this subsample that entered the extension phase. Similarly, at the end of the extension period, MADRS total score mean changes using last observation carried forward (LOCF) for the full analysis set from baseline were also not different between the two groups (Table 3). The percentage of responders (61.9 % v. 55.0%, P= 0.195 and remitters (57.7% v. 52.6%, P = 0.345) on the MADRS scale at the last post-baseline assessment were numerically higher in the AGO-MS group than in the PBO-MS group, but neither comparison reached conventional levels of statistical significance.
Other secondary efficacy measures
There were no significant differences between the groups on, CGI-BP-M depression, mania or general scores at week 8 or month 12. Similarly, no significant differences in change scores on the HRSD-17, Reverse Vegetative Symptoms Scale, or HRSA, LSEQ and QLESQ were observed between the groups at week 8.
Withdrawals
During the 8-week acute treatment period, 86 randomised patients (25.0%) withdrew from the study, 47 as a result of lack of efficacy (22 patients in the AGO-MS group; 25 patients in the PBO-MS group) (Fig.1). Of the 247 patients continuing into the double-blind extension period of the study (125 patients in the AGO-MS group and 122 in the PBO-MS group), a total of 99 were prematurely withdrawn in the two groups; the commonest single reason was lack of efficacy (19 patients in the AGO-MS group; 18 patients in the PBO-MS group) (Fig. 1).
Post hoc analysis
Since the placebo response was very high (61% of patients in the PBO-MS met criteria for response on the MADRS total score), we conducted a post hoc exploratory analyses to examine and understand the impact of placebo response on the results of the trial.
A substantial number of centres had all patients (100%) randomised to PBO-MS meeting the criteria for response at 8 weeks. Therefore, we analysed the data after excluding centres with different rates of placebo response. For these analyses, we included data only from those centres (n = 54) that had randomised at least one patient per treatment group. We then examined the differences on the change from baseline to last post-baseline value at week 8 for MADRS total score between agomelatine and placebo after exclusion of data from centres that had placebo response rates of 100%, ≥90%, ≥80%, etc….0%. Changes in MADRS scores for agomelatine v. placebo groups for these subgroups are displayed in Table 4. Agomelatine was significantly superior to placebo in all these analyses.
Table 4 Post hoc analysis in centres with at least one patient in each group according to different placebo response rates: Montgomery–Åsberg Depression Rating Scale (MADRS) changes in agomelatine and placebo groups, impact of centres with different placebo response rates
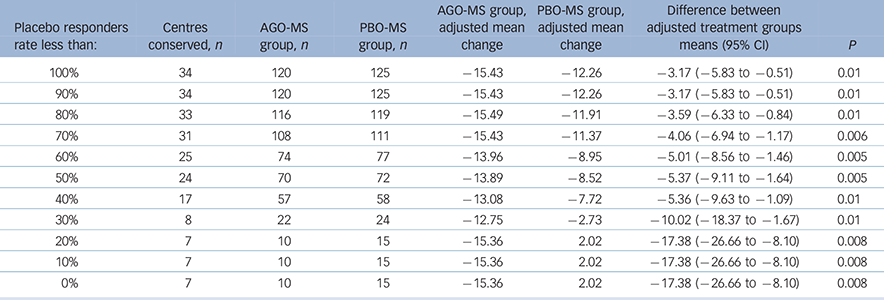
| Placebo
responders rate less than: |
Centres conserved, n |
AGO-MS group, n |
PBO-MS group, n |
AGO-MS
group, adjusted mean change |
PBO-MS
group, adjusted mean change |
Difference
between adjusted treatment groups means (95% CI) |
P |
|---|---|---|---|---|---|---|---|
| 100% | 34 | 120 | 125 | −15.43 | −12.26 | −3.17 (−5.83 to −0.51) | 0.01 |
| 90% | 34 | 120 | 125 | −15.43 | −12.26 | −3.17 (−5.83 to −0.51) | 0.01 |
| 80% | 33 | 116 | 119 | −15.49 | − 11.91 | −3.59 (−6.33 to −0.84) | 0.01 |
| 70% | 31 | 108 | 111 | −15.43 | − 11.37 | −4.06 (−6.94 to −1.17) | 0.006 |
| 60% | 25 | 74 | 77 | −13.96 | −8.95 | −5.01 (−8.56 to −1.46) | 0.005 |
| 50% | 24 | 70 | 72 | −13.89 | −8.52 | −5.37 (−9.11 to −1.64) | 0.005 |
| 40% | 17 | 57 | 58 | −13.08 | −7.72 | −5.36 (−9.63 to −1.09) | 0.01 |
| 30% | 8 | 22 | 24 | −12.75 | −2.73 | −10.02 (−18.37 to −1.67) | 0.01 |
| 20% | 7 | 10 | 15 | −15.36 | 2.02 | −17.38 (−26.66 to −8.10) | 0.008 |
| 10% | 7 | 10 | 15 | −15.36 | 2.02 | −17.38 (−26.66 to −8.10) | 0.008 |
| 0% | 7 | 10 | 15 | −15.36 | 2.02 | −17.38 (−26.66 to −8.10) | 0.008 |
AGO-MS, agomelatine as an add-on to a mood stabiliser; PBO-MS, placebo as an add-on to a mood stabiliser.
Conversely, we were interested in determining if placebo would beat agomelatine if all data were excluded from centres with a particular responder rate to agomelatine (i.e. 100%, ≥90%, ≥80%, etc….0%). Table 5 shows the MADRS change scores for these subgroups. Placebo was not superior to agomelatine in all these analyses except when data from all centres that had ≥30% response were excluded. The number of remaining patients for analyses at this stage was very low.
Table 5 Post hoc analysis in centres with at least one patient in each group according to different agomelatine response rates: Montgomery–Åsberg Depression Rating Scale (MADRS) changes in agomelatine and placebo groups, impact of centres with different agomelatine response rates

| Placebo
responders rate less than: |
Centres conserved, n |
AGO-MS group, n |
PBO-MS group, n |
AGO-MS
group, adjusted mean change |
PBO-MS
group, adjusted mean change |
Difference
between adjusted treatment groups means (95% CI) |
P |
|---|---|---|---|---|---|---|---|
| 100% | 37 | 130 | 132 | −14.68 | −14.65 | −0.03 (−2.59 to 2.54) | 0.98 |
| 90% | 37 | 130 | 132 | −14.68 | −14.65 | −0.03 (−2.59 to 2.54) | 0.98 |
| 80% | 37 | 130 | 132 | −14.68 | −14.65 | −0.03 (−2.59 to 2.54) | 0.98 |
| 70% | 28 | 85 | 61 | −12.23 | −13.26 | 1.04 (−2.21 to 4.28) | 0.53 |
| 60% | 21 | 57 | 61 | −10.47 | −13.17 | 2.70 (−1.18 to 6.59) | 0.17 |
| 50% | 21 | 57 | 61 | −10.47 | −13.17 | 2.70 (−1.18 to 6.59) | 0.17 |
| 40% | 12 | 37 | 43 | −8.38 | −10.96 | 2.58 (−2.13 to 7.28) | 0.27 |
| 30% | 10 | 20 | 23 | −5.67 | −12.11 | 6.43 (1.64 to 11.23) | 0.01 |
| 20% | 10 | 20 | 23 | −5.67 | −12.11 | 6.43 (1.64 to 11.23) | 0.01 |
| 10% | 9 | 15 | 19 | −4.79 | − 11.25 | 6.46 (1.37 to 11.55) | 0.01 |
| 0% | 9 | 15 | 19 | −4.79 | − 11.25 | 6.46 (1.37 to 11.55) | 0.01 |
AGO-MS, agomelatine as an add-on to a mood stabiliser; PBO-MS, placebo as an add-on to a mood stabiliser.
Safety
The percentage of patients in the safety set who reported at least one emergent adverse event on treatment over 8 weeks was numerically higher in the AGO-MS group than in the PBO-MS group (54.4% v. 46.2%) (mean difference: −8.14 (s.e. = 5.37), 95% CI −18.68 to 2.39, P = 0.131). The most frequently affected system organ classes in the AGO-MS group were gastrointestinal disorders (16.4% of patients), nervous system disorders (15.2%), psychiatric disorders (12.3%) and infections and infestations (8.2%), and were similarly reported in the PBO-MS group (17.9%, 16.8%, 12.1%, 9.8%, respectively)
During the 12-month treatment period, the percentage of patients with at least one emergent adverse event showed no relevant difference between the AGO-MS and PBO-MS groups (69.6% v. 64.7%). The most frequent emergent adverse events were headache, nasopharyngitis, depression and somnolence (Table 6). Adverse events were generally less frequently reported in the AGO-MS group than in the PBO-MS group; only nasopharyngitis, and somnolence were more frequently reported in the AGO-MS group than in the PBO-MS group (6.4% v. 4.0%; 5.8% v. 4.0%, respectively).
Table 6 Most frequently reported emergent adverse events a during the 12-month double-blind treatment period (at least 5% of the patients in any group)

| Adverse events | AGO-MS
group (n =171) |
PBO-MS
group (n =173) |
|---|---|---|
| Headache | 8.8 | 11.0 |
| Nasopharyngitis | 6.4 | 4.0 |
| Depression | 5.8 | 7.5 |
| Somnolence | 5.8 | 4.0 |
| Hypomania | 5.3 | 2.9 |
| Nausea | 4.7 | 6.9 |
| Insomnia | 4.1 | 6.4 |
| Dizziness | 4.1 | 5.8 |
| Diarrhoea | 4.1 | 5.2 |
a. Expressed as percent of number of affected patients to number of exposed patients in the considered treatment group.
Manic or hypomanic symptoms
Over the 8-week period, the percentage of patients who experienced manic or hypomanic symptoms was not different between the treatment groups: 4.1% (seven patients: two hypomania, two mania, two elevated mood and one mixed episode) in the AGO-MS group and 3.5% (six patients: three hypomania, two mania, one mood swings) in the PBO-MS group. These symptoms were considered to be treatment related in all patients receiving AGO-MS, and in four of six patients receiving PBO-MS. The mean YMRS total score was stable over the acute period in both treatment groups (change from baseline: −0.38 (s.d. = 3.67) in the AGO-MS group and −0.51 (s.d. = 3.77) in the PBO-MS group). Over the extension period, 13 additional patients (7.6%) in the AGO-MS group and 7 (4.1%) in the PBO-MS group experienced manic or hypomanic symptoms. They were treatment related in all but one patient receiving AGO-MS (one hypomania), and in three of seven patients receiving PBO-MS. Among these symptoms, emergent hypomania was more frequent in the agomelatine group (7/13) than in the placebo group (2/7). The same number of manic episodes (three) was reported in both groups. The mean YMRS total score was stable over the extension period (AGO-MS: 0.49 (s.d. = 5.07); PBO-MS group: 0.25 (s.d. = 4.80)).
Other safety events
One death occurred in the PBO-MS group after 35 days. This death, the result of an upper respiratory tract infection, was considered to be unrelated to the study treatment by the investigator. In both treatment groups, emergent non-fatal serious adverse events were mainly related to psychiatric disorders (including (hypo)manic symptoms) and did not differ between patients in the AGO-MS (n = 12, 7.0%) and PBO-MS (n = 10, 5.8%) groups. Among these events, suicide attempts were less frequent in the AGO-MS group (two patients, 1.2%) than in the PBO-MS group (five patients, 2.9%).
During the 12-month treatment period, no clinically relevant changes or differences between groups over time were detected for serious (non-fatal) or non-serious emergent adverse events leading to treatment withdrawal. Thus, 9 patients, in the AGO-MS group (5.3%) and 8 patients in the PBO-MS group (4.6%) withdrew because of emergent serious adverse event, whereas 28 patients in the AGO-MS group (16.4%) and 25 patients in the PBO-MS group (14.5%) discontinue because of non-serious adverse events.
During the study, no differences between groups over time were detected for biochemistry or haematological parameters, or physical examination including vital signs. No ECG abnormality was recorded. Two patients (n = 1 in the AGO-MS group, n = 1 in the PBO-MS group) had elevations in liver enzyme values more than three times the upper limit of the reference range. Both patients completely recovered.
Discussion
This randomised double-blind placebo-controlled study in patients with bipolar I disorder with current major depressive episode failed to show any beneficial effect of AGO-MS compared with PBO-MS on the primary outcome in the intent-to-treat full analysis sample. The MADRS score changes observed over the acute 8-week period were relatively large (about −15.0) in both treatment groups, and were maintained over the 10-month period. Response (approximately 61%) and remission rates (approximately 53%) on the MADRS scale for both treatment groups were also similar over the acute period. There were similar improvements in both groups on secondary efficacy measures in the full analysis set. Over the double-blind extension period, in the AGO-MS group, numerically but not significantly higher percentages of patients met criteria for response (61.9% v. 55.0%) and remission (57.7% v. 52.6%) compared with the PBO-MS group.
Magnitude of improvement in depression
The magnitude of reduction in depressive symptoms observed in the AGO-MS group in the present study is consistent with the effects noted in the preliminary open study Reference Calabrese, Guelfi and Perdrizet-Chevallier25 but, unexpectedly, a similar magnitude of improvement was also observed in the PBO-MS group. An improvement of 15 points on the MADRS in the PBO-MS group is larger than that observed in previous studies with PBO-MS. In the four large-scale double-blind trials that reported change scores on MADRS or HRSD in the PBO-MS groups, the reductions in depressive symptom scores were 8.06, Reference Nemeroff, Evans, Gyulai, Sachs, Bowden and Gergel12 11.03, Reference van der Loos, Mulder, Hartong, Blom, Vergouwen and de Keyzer35 12.9 Reference Sachs, Nierenberg, Calabrese, Marangell, Wisniewski and Gyulai13 and 13.5. Reference Loebel, Cucchiaro, Silva, Kroger, Sarma and Xu36 The 15-point improvement observed in the PBO-MS group was larger than expected when compared with previous studies and translated to 61% of patients meeting criteria for response in the PBO-MS group. This is also substantially higher than the response rates observed in previous studies (34.9% in Nemeroff et al, Reference Nemeroff, Evans, Gyulai, Sachs, Bowden and Gergel12 31.7% in van der Loos et al, Reference van der Loos, Mulder, Hartong, Blom, Vergouwen and de Keyzer35 27.3% in Sachs et al Reference Sachs, Nierenberg, Calabrese, Marangell, Wisniewski and Gyulai13 and 42% in Loebel et al Reference Loebel, Cucchiaro, Silva, Kroger, Sarma and Xu36 ). Given the challenges experienced in improving depressive symptoms in patients with bipolar depression in real-world clinical practice, such large response rates to PBO-MS in clinical trials are surprising. This finding raises the possibility that higher response rates in the PBO-MS group in the current study may have reduced the power of the study to detect an antidepressant effect of agomelatine.
Placebo response rates and strategies to improve assay sensitivity
High response to placebo has become as significant an issue in bipolar depression trials as in unipolar trials, where the effect was noted longer ago. Reference Walsh, Seidman, Sysko and Gould37 In monotherapy trials, the response to placebo was 26% in the lamotrigine trial Reference Calabrese, Bowden, Sachs, Ascher, Monaghan and Rudd38 and it has steadily increased since then to 55.8% in the recent EMBOLDEN I study. Reference Young, McElroy, Bauer, Philips, Chang and Olausson39 Entry score severity and inflation have been cited as reasons for increased placebo response. In order to address entry score inflation, a cut-off score on the HRSD was used for entry and the improvement in depression was assessed as the change score on MADRS for determination of efficacy. This strategy has not been found to diminish placebo response either in this study or in previous trials. Reference Sachs, Ice, Chappell, Schwartz, Gurtovaya and Vanderburg40,Reference Lombardo, Sachs, Kolluri, Kremer and Yang41 Previous studies suggest that a score of 20 on the HRSD 17-item scale corresponds to a score of 28 on the MADRS. Reference Zimmerman, Posternak and Chelminski42 The mean HRSD score in this study was 25 but the mean MADRS score was only 30, which suggests a potential HRSD score inflation indicating that some patients who were less ill than required by the study protocol may have been recruited into the study.
In a meta-analysis that examined placebo response rates in major depressive disorder, Iovieno & Papakostas Reference Iovieno and Papakostas43 observed that adjunctive studies with placebo response rates >40% were very unlikely to report drug–placebo differences. The present study accords with this conclusion. Previous work has highlighted the remarkable variation in drug–placebo differences by study site (of which there were 67 in the present study). Reference Dunlop, Thase, Wun, Fayyad, Guico-Pabia and Musgnung16 One way to generate an assay-sensitive subgroup from the full analysis set is to exclude from a secondary analysis sites with improbably high response rates. Merlo-Pich et al have suggested this strategy for proof of concept and showed that it reduced the probability of trial failure from around 50% to around 10%. Reference Merlo-Pich, Alexander, Fava and Gomeni15
The objective of clinical trials should be to test new medications in representative clinical populations, which means in populations where the placebo response rates are neither high nor low. Recruitment of such populations has become an increasing problem for trials in psychiatry generally and, therefore, it is important to use secondary analyses to understand the phenomenon of high placebo response. In the present study, there was a substantial number of centres recruiting only a single patient (n = 8/67) and others with 100% response rates to PBO-MS (n = 25/67). Thus, in order to understand the impact of high placebo response on the outcome of this study, we conducted post hoc analyses examining only those centres that randomised at least one patient to each condition and excluding sequentially all patients from centres that had placebo response rates of 100%, ≥90%, ≥80%….etc. Exclusion of patients (both PBO-MS and AGO-MS) from centres that had 100% placebo response yielded results suggesting significantly greater improvements in patients allocated to AGO-MS (P = 0.02). Similarly, AGO-MS was significantly superior to PBO-MS when all patients were excluded from centres that had placebo response rates ≥90%, ≥80%….0% (Table 4). Conversely, when data from centres that had different agomelatine responder rates were excluded, placebo did not beat agomelatine until all centres that had ≥30% response to agomelatine were excluded. At <30%, the data from only 10 out of 67 centres were conserved with a total of only 43 patients. Hence, this analysis is not likely meaningful.
Safety and tolerability
Our findings provide additional data in confirmation of the tolerability and safety profiles of agomelatine 25–50 mg seen in the patients with major depressive disorder and generalised anxiety disorder. Reference Hickie and Rogers19,Reference Stein, Ahokas and De22 No unexpected adverse event was reported. The AGO-MS hepatic side-effects profile was similar to PBO-MS. The rate of emergent adverse effects over long-term treatment was also similar. The occurrence of mania in patients with bipolar disorder with antidepressant treatment, even if they are treated with a mood stabiliser, is a relatively old clinical observation Reference Gijsman, Geddes, Rendell, Nolen and Goodwin44 and a major concern. Reference Valenti, Pacchiarotti, Bonnin, Rosa, Popovic and Nivoli45 Notably, AGO-MS was not associated with a high level of emergence of mania or hypomania symptoms over the study period.
Limitations
This study was conducted in 67 centres in 15 countries, which may have contributed to the large placebo response observed in the study. Such high placebo responses are not consistent with clinical practice in patients with bipolar depression and this raises questions about the quality of centres and the type of patients enrolled into the study. Exclusion of patients from centres with high placebo response rates yielded results that support the efficacy of agomelatine. However, this was based on post hoc analysis and hence, this cannot be taken as firm evidence of the efficacy of agomelatine. Further, in this post hoc analysis, we excluded data from only those centres that had randomised at least one patient to placebo and one patient to agomelatine. One could debate what this minimum number should be and a sensitive analysis combining data-sets from different studies might provide a more precise answer to this question. Although this study used an adjunctive design, refractoriness to mood stabiliser therapy was not prospectively confirmed. It is conceivable that agomelatine may be more effective in patients with bipolar depression who are refractory to mood stabiliser monotherapy.
Implications
The role of antidepressants in acute treatment of bipolar depression continues to remain controversial. In this study, agomelatine was not superior to placebo in improving depressive symptoms based on the changes in primary efficacy measure in acute bipolar depression. In general, there is a paucity of trial data on both the short- and long-term use of antidepressants in patients with bipolar disorder, so there is a real need to maximise understanding of the data that does exist. Post hoc analysis excluding patients from centres with a high placebo response suggested efficacy for agomelatine. We believe this procedure to be highly informative, compared with the publication of a mean response rate. The failure in this case to demonstrate benefit in the full analysis set underlines the challenge for improving the quality of patient recruitment to minimise placebo response. Failing that, one way to increase the assay sensitivity might be to set a priori criteria for excluding centres with certain rates of placebo response. A sensitivity analysis combining data from a number of bipolar depression studies might provide a better answer to what the ‘cut-off’ should be. Given that the average placebo response rates have not been higher than 62% in previous bipolar depression studies, a more stringent cut-off of 70% or greater might be appropriate if this is supported by a sensitivity analysis. Given the paucity of treatments, this may be particularly important for the clinical development of treatments for bipolar depression.
Acknowledgements
The Agomelatine Study Group includes the following national coordinators: S. Strejilevitch (Buenos Aires, Argentina), G. Johnson (Greenwich, Australia), S. Kasper (Wien, Austria), F. Kapczinski (Porto Alegre, Brazil), L. N. Yatham (Vancouver, Canada), P. Bech (Hillerod, Denmark), A. Ahokas (Helsinki, Finland), M. Bourin (Nantes, France), H. Grünze (Munich, Germany), R. Sagar (Dehli, India), Kyoo-Seob Ha (Gyeonggi-do, Korea), W. A. Nolen (Groningen, The Netherlands), J. K. Rybakowski (Poznan, Poland), R. Emsley (Tygerberg, South Africa), E. Vieta (Barcelona, Spain). M. Bourin (Nantes, France), J. R. Calabrese (Cleveland, USA), E. Frank (Pittsburgh, USA), G. M. Goodwin (Oxford, UK), G. Johnson (Greenwich, Australia) E. Vieta (Barcelona, Spain) L. Yatham (Vancouver, Canada) were scientific advisors for the study.








eLetters
No eLetters have been published for this article.