


Cardiovascular disease (CVD) is a leading cause of morbidity and mortality, Reference Mathers and Loncar1 and its economic impact exceeds that of other diseases. Reference Roger, Go, Lloyd-Jones, Benjamin, Berry and Borden2 Substantial achievements have been made in the reduction of CVD mortality rates, by decreasing exposure to traditional risk factors such as smoking and hypertension. Reference Ford, Ajani, Croft, Critchley, Labarthe and Kottke3–Reference Vartiainen, Laatikainen, Peltonen, Juolevi, Mannisto and Sundvall5 Researchers have argued that psychosocial factors are as important as traditional risk factors in preventing the onset of CVD. Reference Yusuf, Hawken, Ounpuu, Dans, Avezum and Lanas6 In a previous meta-analysis summarising 20 studies, anxiety was associated with a 26% increased risk of incident coronary heart disease (CHD), and a 48% increased risk of cardiac death, after adjustment for demographic and biological risk factors and health behaviours. Reference Roest, Martens, de Jonge and Denollet7 Since publication of that review in 2010, various new studies have been published; these justify an update of the previous meta-analysis. Several of the new studies examined anxiety disorders instead of self-reported anxiety symptoms, which is what the majority of earlier studies were based on. In addition, three aspects require a closer examination. First, the previous meta-analysis was restricted to CHD; Reference Roest, Martens, de Jonge and Denollet7 inclusion of similar expressions of atherosclerotic vascular disease in the brain (stroke) or in the periphery (peripheral vascular disease) would provide a more complete picture of the association between anxiety and CVD. Second, since depression is highly comorbid with anxiety, Reference Lamers, van Oppen, Comijs, Smit, Spinhoven and van Balkom8 and is associated with CVD onset and progression, Reference Nicholson, Kuper and Hemingway9 it has to be ascertained whether anxiety is a true risk factor for CVD – independent of depression – by considering studies that explicitly adjust for or exclude co-occurring depression. The publication of new studies has allowed these analyses. Third, quantifying the association between anxiety and the onset of CVD does not corroborate a causal association. Ascertaining causality is essential when aiming to prevent CVD. In 1965 Hill formulated criteria for causality, Reference Hill10 but these had not been evaluated explicitly regarding the anxiety–CVD association. We conducted a meta-analysis of prospective population-based studies in persons free from CVD to examine whether anxiety (anxiety disorder or increased anxiety symptoms) was associated with the onset of atherosclerotic CVD, compared with no anxiety (no anxiety disorder or no/minimal anxiety symptoms). Reference Moher, Liberati, Tetzlaff and Altman11 Secondary outcomes included CHD, myocardial infarction, stroke and cardiovascular mortality. Moreover, we reviewed whether Hill's criteria for causality were being met, by conducting relevant subgroup analyses and qualitatively reviewing additional literature.
Method
We searched without language restrictions in PubMed, EMBASE and PsycINFO (from inception up to 28 October 2013) for prospective population-based studies that examined whether anxiety (anxiety disorder or increased anxiety symptoms) was associated with the new onset of atherosclerotic CVD, relative to no anxiety (no anxiety disorder or no/minimal anxiety symptoms) in humans free from CVD. In these searches we combined words indicating anxiety, CVD and prospective design. Both text and keywords were used (see online supplement DS1).
Two raters (N.M.B. and A.S.) independently screened titles and abstracts and assessed for eligibility the full text of part of the studies. The following inclusion criteria were applied:
-
(a) longitudinal design, with the anxiety assessment prior to assessment of CVD;
-
(b) assessment of new-onset atherosclerotic CVD (studies not excluding participants with CVD at baseline were allowed if adjusted for baseline CVD status);
-
(c) comparison of CVD risk between individuals with and without anxiety.
The assessment of anxiety could be based on self-reporting of symptoms, a clinical diagnostic assessment or registration in a managed care database. Consequently, both anxiety symptoms and anxiety disorders were included. Atherosclerotic CVD included CHD, myocardial infarction, ischaemic heart disease, cardiovascular mortality, sudden cardiac death, stroke and peripheral arterial disease, or a combination of these end-points. As differentiation between angina pectoris and non-cardiac chest pain might be difficult, angina was not included in our definition of CVD, yet allowed when part of a broader outcome definition. Likewise, we restricted the analyses to atherosclerotic CVD, yet allowed heart failure when part of a broader outcome definition. Articles not presenting original data or consisting only of abstracts were excluded. If multiple papers reported similar outcomes based on the same data-set, we chose the study with the largest sample or longest follow-up interval. Consensus was reached in cases of initial disagreement.
Data extraction
Three authors (N.M.B., M.B. and A.S.) independently extracted descriptive information from each study using a predesigned collection form. This information included characteristics of the study population (gender, mean age, age range), publication year, study location (continent), follow-up duration, anxiety measurement, type of CVD outcome, CVD status at baseline, covariate adjustment, baseline depression status and effect sizes with confidence intervals. If a single study reported several anxiety measures, we included the dimension that best reflected general psychological anxiety. If anxiety was classified into more than two categories, the category with highest anxiety was compared with the category with lowest anxiety. Meta-analyses were performed for the main outcome (CVD) and performed separately for the predefined secondary outcomes. If studies examined multiple outcomes in the same data-set, the broadest was used as the main outcome. If applicable, other outcomes were allowed to be included in a meta-analysis of secondary outcomes. Studies could therefore be included in more than one meta-analysis. If risk estimates were provided for subgroups (for example, men and women), associations are reported for these subgroups with their corresponding sample sizes.
Risk of bias
Studies may differ in quality, which may bias results. To assess the risk of bias, all studies were scored for six domains relevant to prognostic studies: Reference Hayden, Cote and Bombardier12 study participation, study attrition, prognostic factor measurement, outcome measurement, confounding, statistical analysis and reporting. A summary score was computed (online Table DS1).
Statistical analysis
Meta-analyses were performed for the main outcome of CVD to assess whether anxiety increases risk irrespective of the location in the body and also to find secondary outcomes (CHD, myocardial infarction, stroke, cardiovascular mortality, peripheral vascular disease). Hazard ratios (HRs) were used as the common measure of associations; relative risks (RRs) and odds ratios (ORs) were considered equivalent. Since we expected considerable heterogeneity across studies, HRs were pooled using the random effects model. Forest plots were produced to enable visual assessment of the HRs and corresponding 95% confidence intervals. Heterogeneity of HRs was evaluated by the Cochrane Q statistic and the I 2 statistic. Reference Higgins, Thompson, Deeks and Altman13 The possibility of publication bias was evaluated using the Egger test of the intercept and visual inspection of a funnel plot. Reference Egger, Smith, Schneider and Minder14 The Duval & Tweedie non-parametric trim and fill procedure was used to adjust for publication bias. Reference Duval and Tweedie15 For the main outcome the risk of bias across studies was further explored by removing one study at a time and reanalysing the remaining data-set. Additionally, we investigated the influence of selected characteristics on the pooled estimate for CVD. Prespecified subgroup analyses were conducted for the following categorical variables: gender; age group (mean age of cohort <55 years, ⩾65 years, ‘other’); continent (North America, Europe, Asia); anxiety type (general, panic, post-traumatic stress); anxiety severity (disorder v. symptoms); sample CVD-free at baseline (yes v. no, but adjusted for baseline CVD); adjustment for gender, age and at least one other traditional CVD risk factor such as smoking, physical activity, hypertension, body mass index (yes v. no); and exclusion of people with depression or adjustment for co-occurring depression (yes v. no). For the age group variable, if the mean age was not reported the age range was used, and if this could not be classified into <55 or ⩾65 years it was classified as ‘other’. Meta-regression was used to study the impact of the continuous characteristics duration of follow-up, year of publication and study quality on the pooled estimates. All analyses were performed with Comprehensive Meta-Analysis version 2.2.064. Reference Biostat16
Criteria of causality
Hill's criteria of causality include strength, consistency, specificity, temporality, biological gradient, plausibility, coherence, experiment and analogy (online Table DS2). The more criteria that are met, the more likely it is that an association is causal. We used these criteria to examine the likelihood of causality in the association between anxiety and new-onset CVD. Reference Hill10 Several of these criteria can be evaluated by the results of the meta-analysis and corresponding subgroup analyses. Other criteria require a qualitative review of the literature, for which we additionally searched for relevant studies (Table DS2).
Results
The literature search resulted in 2489 records (Fig. 1). Of these, 86 full-text articles were assessed for eligibility. Nine studies were excluded because they used the same data-set with similar outcomes, but with smaller sample sizes or shorter follow-up periods, Reference Chen, Hu, Lee and Lin17–Reference Haynes, Feinleib and Kannel23 two of which were also in Russian. Reference Gafarov, Gromova, Gagulin and Gafarova24,Reference Gafarov, Pak, Gagulin and Gafarova25 Three authors provided measures of association on request; Reference Davidson, Mostofsky and Whang26–Reference Holt, Phillips, Jameson, Cooper, Dennison and Peveler28 three studies were excluded because a continuous anxiety measure was used and dichotomised associations were not provided. Reference Shirom, Toker, Jacobson and Balicer29–Reference Eaker, Sullivan, Kelly-Hayes, D'Agostino and Benjamin31 In total, 37 papers were included reporting 62 comparisons (online Table DS3). All of these papers were published between 1991 and 2013. The number of participants per study ranged from 506 to 404 643, and the average follow-up ranged from 1 year to 24 years. Nine studies assessed anxiety diagnoses and 25 studies assessed anxiety symptoms. In two studies, estimates were provided only for subgroups of participants: in one, the sample was restricted to women, Reference Carriere, Ryan, Norton, Scali, Stewart and Ritchie32 and in the other to people of Black ethnicity. Reference Brenes, Kritchevsky, Mehta, Yaffe, Simonsick and Ayonayon33 Only one study addressed peripheral arterial disease; Reference Bowlin, Medalie, Flocke, Zyzanski and Goldbourt34 a separate meta-analysis was thus not possible for this outcome.
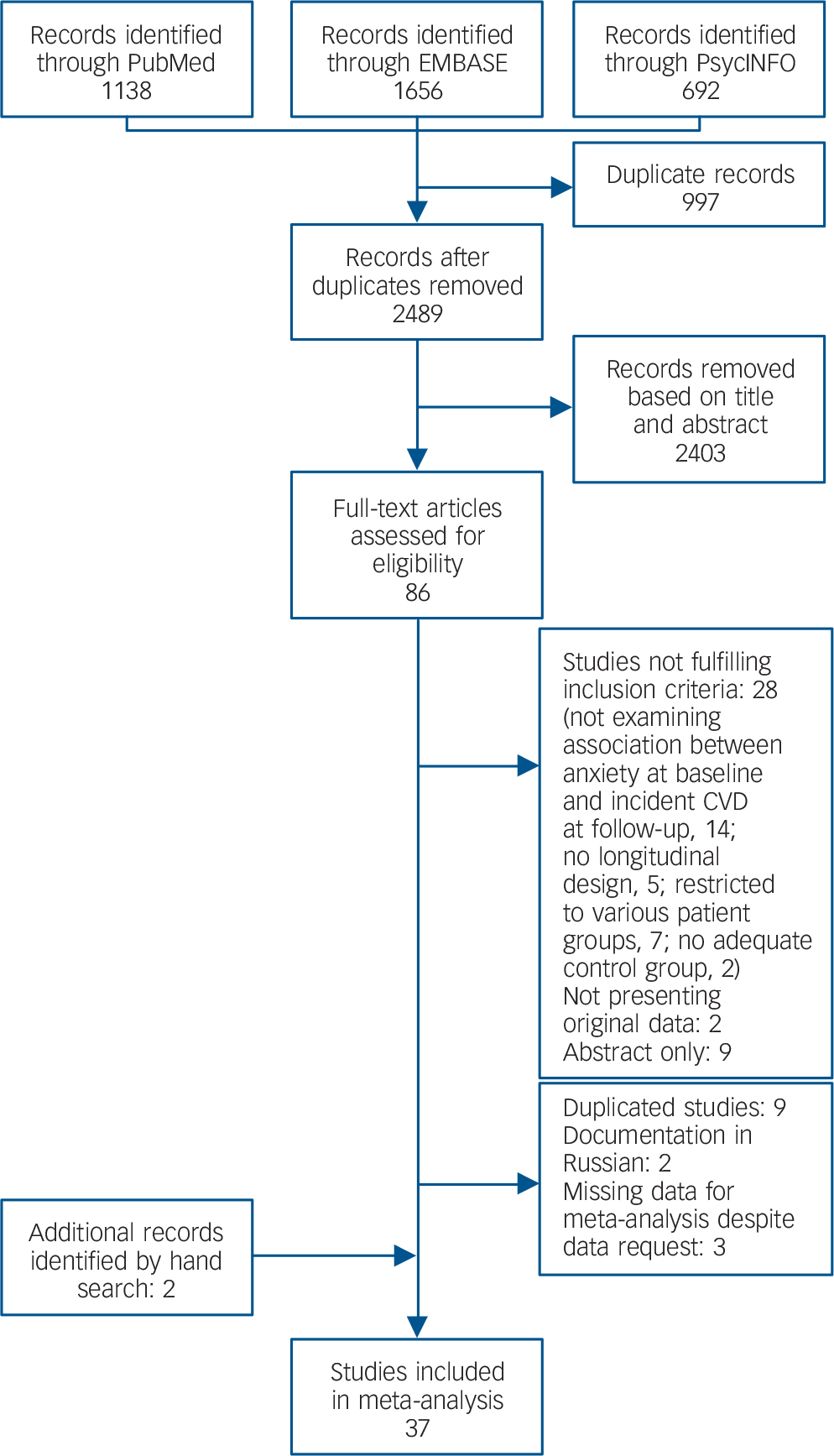
Fig. 1 Selection of studies.
Meta-analyses
Pooled analysis revealed that anxiety was associated with a 52% increased incidence of CVD (HR = 1.52, 95% CI 1.36–1.71) based on a total of 1 565 699 persons (Fig. 2). We detected strong evidence of heterogeneity (Q = 483.7, P<0.001; I 2 = 92.1). Reference Higgins, Thompson, Deeks and Altman13 Subsequent removal of each study from the analyses had little impact on the overall estimate, except for elimination of Gomez-Caminero et al's data, Reference Gomez-Caminero, Blumentals, Russo, Brown and Castilla-Puentes35 which gave a pooled HR of 1.44 (95% CI 1.33–1.57) and reduced the heterogeneity (Q = 124.5, P<0.001; I 2 = 70.3). Egger's test for publication bias was significant (one-tailed P = 0.03), indicating that publication bias was likely (online Fig. DS1). After adjustment for publication bias the pooled HR dropped 21% but remained significant (HR = 1.41, 95% CI 1.26–1.57). Anxiety was also significantly related to all secondary outcomes, with HRs varying from 1.38 for myocardial infarction (95% CI 1.16–1.64) to 1.74 for stroke (95% CI 1.25–2.43) (Table 1). Thus, anxiety increased the risk of atherosclerotic CVD irrespective of its location in the body. Publication bias was likely for all outcomes except myocardial infarction (Egger's tests of the intercept one-tailed P-values for myocardial infarction, CHD, cardiovascular mortality and stroke were 0.12, 0.03, <0.001 and 0.05 respectively). Adjustment for publication bias reduced the strength of the associations but they remained significant (data not shown). None of the subgroup analyses or meta-regression analyses demonstrated any difference in size of the association between anxiety and new-onset CVD (Table 2). Results are therefore consistent across studies examining anxiety symptoms and disorders, and across studies that considered co-occurring depression and those that did not.

Fig. 2 Forest plot.
Table 1 Meta-analyses of association of anxiety with primary and secondary outcomes
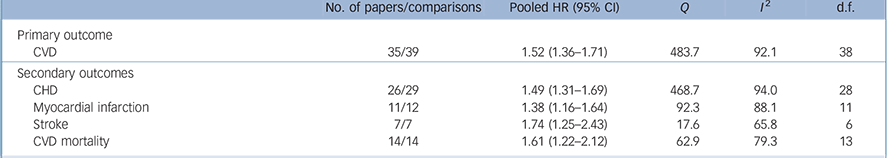
| No. of papers/comparisons | Pooled HR (95% CI) | Q | I 2 | d.f. | |
|---|---|---|---|---|---|
| Primary outcome | |||||
| CVD | 35/39 | 1.52 (1.36–1.71) | 483.7 | 92.1 | 38 |
| Secondary outcomes | |||||
| CHD | 26/29 | 1.49 (1.31–1.69) | 468.7 | 94.0 | 28 |
| Myocardial infarction | 11/12 | 1.38 (1.16–1.64) | 92.3 | 88.1 | 11 |
| Stroke | 7/7 | 1.74 (1.25–2.43) | 17.6 | 65.8 | 6 |
| CVD mortality | 14/14 | 1.61 (1.22–2.12) | 62.9 | 79.3 | 13 |
CHD, coronary heart disease; CVD, cardiovascular disease; HR, hazard ratio.
Table 2 Subgroup analyses for the primary outcome of cardiovascular disease
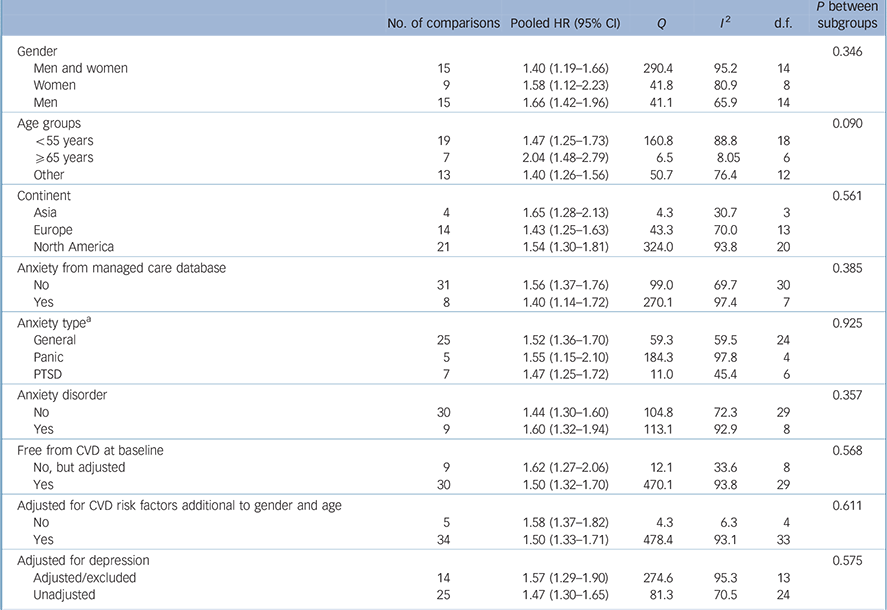
| No. of comparisons | Pooled HR (95% CI) | Q | I 2 | d.f. |
P between subgroups |
|
|---|---|---|---|---|---|---|
| Gender | 0.346 | |||||
| Men and women | 15 | 1.40 (1.19–1.66) | 290.4 | 95.2 | 14 | |
| Women | 9 | 1.58 (1.12–2.23) | 41.8 | 80.9 | 8 | |
| Men | 15 | 1.66 (1.42–1.96) | 41.1 | 65.9 | 14 | |
| Age groups | 0.090 | |||||
| <55 years | 19 | 1.47 (1.25–1.73) | 160.8 | 88.8 | 18 | |
| ⩾65 years | 7 | 2.04 (1.48–2.79) | 6.5 | 8.05 | 6 | |
| Other | 13 | 1.40 (1.26–1.56) | 50.7 | 76.4 | 12 | |
| Continent | 0.561 | |||||
| Asia | 4 | 1.65 (1.28–2.13) | 4.3 | 30.7 | 3 | |
| Europe | 14 | 1.43 (1.25–1.63) | 43.3 | 70.0 | 13 | |
| North America | 21 | 1.54 (1.30–1.81) | 324.0 | 93.8 | 20 | |
| Anxiety from managed care database | 0.385 | |||||
| No | 31 | 1.56 (1.37–1.76) | 99.0 | 69.7 | 30 | |
| Yes | 8 | 1.40 (1.14–1.72) | 270.1 | 97.4 | 7 | |
| Anxiety type a | 0.925 | |||||
| General | 25 | 1.52 (1.36–1.70) | 59.3 | 59.5 | 24 | |
| Panic | 5 | 1.55 (1.15–2.10) | 184.3 | 97.8 | 4 | |
| PTSD | 7 | 1.47 (1.25–1.72) | 11.0 | 45.4 | 6 | |
| Anxiety disorder | 0.357 | |||||
| No | 30 | 1.44 (1.30–1.60) | 104.8 | 72.3 | 29 | |
| Yes | 9 | 1.60 (1.32–1.94) | 113.1 | 92.9 | 8 | |
| Free from CVD at baseline | 0.568 | |||||
| No, but adjusted | 9 | 1.62 (1.27–2.06) | 12.1 | 33.6 | 8 | |
| Yes | 30 | 1.50 (1.32–1.70) | 470.1 | 93.8 | 29 | |
| Adjusted for CVD risk factors additional to gender and age | 0.611 | |||||
| No | 5 | 1.58 (1.37–1.82) | 4.3 | 6.3 | 4 | |
| Yes | 34 | 1.50 (1.33–1.71) | 478.4 | 93.1 | 33 | |
| Adjusted for depression | 0.575 | |||||
| Adjusted/excluded | 14 | 1.57 (1.29–1.90) | 274.6 | 95.3 | 13 | |
| Unadjusted | 25 | 1.47 (1.30–1.65) | 81.3 | 70.5 | 24 | |
CVD, cardiovascular disease; HR, hazard ratio; PTSD, post-traumatic stress disorder.
a. Only two studies assessed phobia; these were removed for this subgroup analysis.
Causality criteria
Strength
The strength of associations varied widely between studies (Fig. 2). The pooled HR for people with anxiety to develop CVD was 1.52 (95% CI 1.36–1.71). After taking publication bias into account this ratio attenuated to 1.41 (95% CI 1.26–1.57). This risk estimate might be an overestimation because confounders were insufficiently accounted for: for example, only a third of studies adjusted for comorbid depression, which is regarded as a CVD risk factor, Reference Nicholson, Kuper and Hemingway9 and only a minority of studies adjusted for psychotropic medication, which some have associated with increased cardiovascular risk. Reference Cohen, Gibson and Alderman36–Reference Lapane, Zierler, Lasater, Barbour, Carleton and Hume39 Some confounders were only assessed globally, and changes in exposure to confounders over time were almost never accounted for, although they might have influenced the association (see post-hoc analysis). Reference Albert, Chae, Rexrode, Manson and Kawachi40 The presence of depression at a later time particularly requires attention, since depression may develop as a comorbid condition thus complicating anxiety. Contrarily, since the exact pathways by which anxiety causes CVD are unknown, risk estimates might be underestimated because confounding variables such as hypertension or dyslipidaemia could have been mediators. Results of the meta-analyses, however, indicate that anxiety is an independent risk factor, because risk estimates from studies that accounted for depression (14 comparisons; HR = 1.57, 95% CI 1.29–1.90) or multiple CVD risk factors (34 comparisons; HR = 1.50, 95% CI 1.33–1.71) were comparable with the pooled HR.
Consistency
Studies to date are heterogeneous regarding gender and age of population, country of investigation, duration of follow-up and year of publication (Table DS3). However, these characteristics did not significantly modify the association between anxiety and new-onset CVD (Tables 2 and 3), indicating that this association is rather consistent. In contrast to previous results, Reference Batelaan, ten Have, van Balkom, Tuithof and de Graaf41 we found the association between anxiety and new-onset CVD to be similar for all anxiety types.
Table 3 Meta-regression for the primary outcome a

| No. of papers/comparisons | B (95% CI) | P | |
|---|---|---|---|
| Duration of follow-up | 35/39 | 0.0103 (−0.0084 to 0.0267) | 0.22 |
| Year of publication | 35/39 | −0.0020 (−0.0211 to 0.0171) | 0.84 |
| Quality | 35/39 | −0.0386 (−0.1134 to 0.0362) | 0.31 |
a. Meta-regression was performed using moments of methods in Comprehensive Meta-analysis 2.
Specificity
Anxiety has been associated with various somatic diseases in both cross-sectional and longitudinal studies. Reference Sareen, Cox, Clara and Asmundson42–Reference Nicholl, Halder, Macfarlane, Thompson, O'Brien and Musleh45 We found an association between anxiety and cardiovascular mortality, whereas previous studies showed mixed results for associations between anxiety and overall mortality. Reference Laan, Termorshuizen, Smeets, Boks, de Wit and Geerlings46–Reference Mykletun, Bjerkeset, Dewey, Prince, Overland and Stewart49 However, as acknowledged by Hill, a less than highly specific association can still reflect causality. For example, a possible hypothesis is that anxiety causes dysregulation of homoeostatic mechanisms, which may result in inflammation (i.e. a specific result), and contribute to the development of various somatic diseases.
Temporality
To ascertain that anxiety precedes CVD, associations must be prospectively studied in CVD-free participants. Exclusion of studies that adjusted for pre-existing CVD did not alter the risk estimates (HR = 1.50, 95% CI 1.32–1.70; see Table 2). Reference Phillips, Batty, Gale, Deary, Osborn and MacIntyre27,Reference Carriere, Ryan, Norton, Scali, Stewart and Ritchie32,Reference Brenes, Kritchevsky, Mehta, Yaffe, Simonsick and Ayonayon33,Reference Mykletun, Bjerkeset, Dewey, Prince, Overland and Stewart49–Reference Yasuda, Mino, Koda and Ohara54 However, exclusion of people with CVD at baseline cannot rule out the presence of subclinical CVD, by which causation in the opposite direction remains an option. Stronger evidence for a temporal relationship running from anxiety to CVD came from one study. It followed a young cohort – in which the presence of subclinical CVD is unlikely – for 37 years, and found anxiety to be associated with incident CVD. Reference Janszky, Ahnve, Lundberg and Hemmingsson55 Still, low cardiovascular fitness at age 18 years in the absence of subclinical CHD might advance the onset of depression in later years. Reference Aberg, Waern, Nyberg, Pedersen, Bergh and Aberg56 De Jonge & Roest postulated a bidirectional hypothesis between depression and CVD, i.e. ‘mutually reinforcing disorders’. Reference De Jonge and Roest57 Additionally, an unknown underlying factor might cause both anxiety and (probably at a slower pace) CVD, for example a common genetic vulnerability. To our knowledge, this latter hypothesis has not yet been examined.
Biological gradient
If causal, higher exposure to anxiety (i.e. greater severity, longer duration) increases the CVD risk. Risk estimates for anxiety disorders (9 studies; HR = 1.60, 95% CI 1.32–1.94) v. symptoms (25 studies; HR = 1.44, 95% CI 1.30–1.60) were higher, but not significantly so (Table 2). Thus, clear evidence for a biological gradient is lacking. In the reviewed papers, a dose–response relationship based on anxiety duration was globally investigated by Nicholson et al, Reference Nicholson, Fuhrer and Marmot58 who stratified the risk of CHD events across those with former, persistent or new distress or anxiety. Their results did not support a biological gradient either. A more detailed assessment on the amount of exposure, for example with anxiety assessed on a continuous scale, or with life chart interviews assessing duration and severity of anxiety over time, might be needed to reveal a biological gradient, if present.
Plausibility
The leading cause of CVD is atherosclerosis, which can be enhanced both indirectly by behavioural factors and directly by physiological processes. Some evidence exists that these pro-atherogenic factors are associated with anxiety. For example, anxiety disorders have been associated with smoking, lower physical activity and poor diet. Reference Moylan, Jacka, Pasco and Berk59–Reference Jacka, Mykletun, Berk, Bjelland and Tell61 In addition, anxiety has been associated with metabolic abnormalities such as increased cholesterol levels and hypertension. Reference Papakostas, Ongur, Iosifescu, Mischoulon and Fava62,Reference Player and Peterson63 Activation of the immune system has also been related to anxiety, Reference Pitsavos, Panagiotakos, Papageorgiou, Tsetsekou, Soldatos and Stefanadis64 as has a hypercoagulable state. Reference Von Kanel, Mills, Fainman and Dimsdale65 Regarding hypothalamic–pituitary–adrenal (HPA) axis functioning, evidence is ambiguous. Reference Young, Abelson, Liberzon, Blanchard, Griebel and Nutt66,Reference Vreeburg, Zitman, van Pelt, Derijk, Verhagen and van Dyck67 Furthermore, anxiety is thought to be marked by an aberrant autonomic heart control with elevated sympathetic activity, Reference Friedman68 although this might be driven by antidepressants. Reference Licht, Penninx and de Geus69 In summation, various pro-atherogenic factors are more frequently found in individuals with anxiety, which concurs with the currently accepted understanding of pathophysiological processes in CVD.
Coherence
Data from animal studies could provide material for the coherence criterion, although studies examining anxiety and CVD are lacking. However, some animal studies have investigated stress-related social factors in association with pathophysiological processes leading to CVD. A review of 14 studies among monkeys concludes that there is some evidence for an association between stress due to disruption of dominance hierarchy and coronary atherosclerosis. Reference Petticrew and Davey Smith70 Another study in monkeys found that social separation led to altered autonomic activity (higher heart rates), which may exacerbate atherosclerosis. Reference Watson, Shively, Kaplan and Line71 A study of mice found that highly anxious mice showed greater chronic stress burden (corticosterone levels) and suppressed protective immunity (levels of inflammatory agents) than mice with low anxiety levels. Reference Dhabhar, Saul, Holmes, Daugherty, Neri and Tillie72 In summation, there is some indication for the criterion of coherence, but convincing evidence is lacking.
Experiment
Primary prevention studies investigating whether effective anxiety treatment indeed prevents CVD onset have not been conducted, and would present quite a challenge. Some small studies that investigated the effect of anxiety treatment on the postulated biological pathways have observed potential cardioprotective effects. For example, treatment with selective serotonin reuptake inhibitors (SSRIs) normalised HPA axis dysregulation, Reference Lenze, Mantella, Shi, Goate, Nowotny and Butters73 and biofeedback, cognitive–behavioural therapy and/or antidepressant medication improved autonomic nervous system functioning. Reference Rice, Blanchard and Purcell74–Reference Sullivan, Kent, Kleber, Martinez, Yeragani and Gorman76 In contrast, antidepressants have also been shown to impair autonomic nervous system activity. Reference Licht, Penninx and de Geus69,Reference Licht, de Geus, van Dyck and Penninx77 Moreover, whether effects on biomarkers can be translated to the onset of CVD remains to be seen. Studies investigating the prevention of cardiovascular events by treating anxiety in patients with CVD (i.e. secondary prevention) have not been carried out either. Such studies sound appealing to start with because of the well-defined high-risk group. However, it is unknown whether the underlying pathogenic mechanisms of anxiety are similar in people with or without CVD. In addition, for depression, a few large-scale secondary prevention studies have generally found no evidence that the treatment of depression improves cardiac outcome. Reference Thombs, de Jonge, Coyne, Whooley, Frasure-Smith and Mitchell78 The authors expounded on these disappointing results by arguing that effect sizes of antidepressant treatment were too low to expect cardioprotective effects. In line with this argument, secondary analyses have shown that people who do not respond to antidepressant treatment are particularly highly at risk of adverse CVD outcomes. Reference De Jonge, Honig, van Melle, Schene, Kuyper and Tulner79,Reference Carney, Blumenthal, Freedland, Youngblood, Veith and Burg80
Analogy
Other psychosocial factors have been postulated to increase the risk of CVD. Whereas the association between hostility and cardiovascular functioning appeared to be mainly driven by covariates, Reference Chida and Steptoe81 the adjusted HR for depression and CVD onset was 1.81. Reference Nicholson, Kuper and Hemingway9 Although we consider depression to be an analogous case, interpretation of the depression–CVD association is, like the anxiety–CVD association, hampered by publication bias, the possibility of reverse causation and residual confounding.
Discussion
A total of 37 studies, with 1 565 699 participants, were included in the meta-analysis on anxiety and CVD. This is almost twice the number of studies included in the previous meta-analysis focusing on CHD, Reference Roest, Martens, de Jonge and Denollet7 thereby justifying an update. Anxiety significantly increased the hazard ratio for incident atherosclerotic CVD by 52%. Although there was evidence of publication bias, imputing potentially missing studies resulted in an HR of 1.41 for the onset of CVD. The association between anxiety and new-onset CVD thereby appears to be at least of similar strength to that between well-established risk factors and CVD. For example, meta-analytic data have shown that the adjusted RRs for CVD associated with moderate overweight and obesity were 1.17 (95% CI 1.11–1.23) and 1.49 (95% CI 1.32–1.67) respectively, Reference Bogers, Bemelmans, Hoogenveen, Boshuizen, Woodward and Knekt82 and that the metabolic syndrome is associated with an increased risk of cardiovascular events or death (adjusted pooled RR = 1.54, 95% CI 1.32–1.79). Reference Gami, Witt, Howard, Erwin, Gami and Somers83 In addition, a high level of physical activity protected against CVD both in men (pooled RR = 0.76, 95% CI 0.70–0.82) and in women (pooled RR = 0.73, 95% CI 0.68–0.78). Reference Li and Siegrist84
In the introduction we mentioned three aspects requiring additional research. First, it was unknown whether anxiety induces vascular abnormalities independent of their localisation along the arterial tree: in the brain (stroke), the heart (CHD, myocardial infarction) or the periphery. We therefore included all outcomes of atherosclerotic vascular disease. Anxiety increased the risk of all secondary outcomes (CHD, myocardial infarction, cardiovascular mortality, stroke); there is therefore no reason to assume that different mechanisms are causing CVD in specific locations only. Peripheral vascular disease was not investigated as a secondary outcome, since this category included only one study. Further research is needed to verify the risk of new-onset peripheral vascular disease associated with anxiety. Second, the extent to which anxiety increases CVD risk, independent of co-occurring depression, required further research. Unlike previous research, the 14 studies in our meta-analysis adjusted for comorbid depression or excluded people with depression. Subgroup analyses based on whether studies considered co-occurring depression revealed similar risk estimates, indicative of the independence of anxiety as a cardiovascular risk factor. However, depression occurring at a later time has not been assessed in studies to date, and could not be considered. The risk estimate for anxiety in our meta-analysis was not very different from the one in the meta-analysis for depression (e.g. Nicholson et al), Reference Nicholson, Kuper and Hemingway9 suggesting that the effects of anxiety and depression are comparable. Results thus underline the need to adjust for anxiety when examining the impact of depression. As most studies on depression have not included anxiety as a covariate, the impact of depression may even have been overestimated (e.g. Nicholson et al). Reference Nicholson, Kuper and Hemingway9 In the light of all studies examining psychosocial factors and the onset of CVD, one might argue that, rather than anxiety or depression, a non-specific ‘umbrella’ factor such as general distress, neuroticism or negative affect accounts for the increased risk of CVD. Third, criteria for causal inference in the anxiety–CVD association had not previously been systematically evaluated. We used the meta-analysis, several subgroup analyses and an additional critical review of the literature to evaluate whether Hill's criteria for causality were met. Results to date largely argue in favour of a causal relationship. However, there is no clear evidence yet for coherence, analogy, a biological gradient or experimental data.
Strengths and limitations
An important strength of this study is the integration of a quantitative summary (meta-analysis) and a qualitative assessment (application of Hill's criteria for causality). Several limitations should be acknowledged as well. First, a high degree of heterogeneity was observed in the studies included. We pooled studies that differed in sample characteristics, anxiety assessment, CVD outcome assessment and statistical analysis. Subgroup analyses, however, suggested that the pooled estimates between these characteristics did not differ. Second, we restricted our analyses to atherosclerotic CVD, therefore no conclusion can be provided with respect to (for example) heart failure. Third, studies differed in the extensiveness of adjustment to covariates and the accuracy by which covariates were measured. This was addressed in the assessment of bias. Meta-regression analysis of the risk of bias scores was not significant.
Study implications
Anxiety may be a point of particular interest for the prevention of CVD incidence. Anxiety is associated with an increased risk of CVD, and this risk appears to be independent of traditional risk factors and depression. In addition, its magnitude is comparable with that of traditional risk factors. Moreover, the prevalence of anxiety disorders and symptoms is high, Reference Kessler, Chiu, Demler, Merikangas and Walters85–Reference Lewinsohn, Shankman, Gau and Klein88 effective treatments reducing the exposure to anxiety as a risk factor are available, 89 and there is significant potential to increase the number of people being treated. Reference Wang, Lane, Olfson, Pincus, Wells and Kessler90 Applying Hill's criteria, our study largely argues in favour of causality, but has revealed important gaps in knowledge, particularly experimental evidence. Deriving experimental evidence from primary prevention studies would present quite a challenge. To begin with, secondary prevention studies might be more feasible. However, before embarking on large-scale experimental programmes it seems sensible to examine in more detail the biological and behavioural underpinnings of the association between anxiety and CVD. By doing so, targets for intervention might be identified that would decrease anxiety and subsequently the magnitude of the worldwide CVD health problem. Until such time that a better insight is gained into the underlying mechanisms, the best advice appears to be to focus on general (behavioural) cardiovascular risk reduction in high-risk populations. As to the primary target of risk intervention, the American Heart Association has stated that ‘adoption of healthy life habits remains the cornerstone of primary prevention’. Reference Pearson, Blair, Daniels, Eckel, Fair and Fortmann111
Acknowledgements
We thank Caroline Planting of the VU University library for her help with the literature search. We also thank our colleague researchers Richard Holt, Anna Phillips and Karina Davidson for providing additional information regarding their studies.







eLetters
No eLetters have been published for this article.